
L.S. Schneider1, D.A. Bennett2, M.R. Farlow3, E.R. Peskind4, M.A. Raskind4, M. Sano5, Y. Stern6, S. Haneline7,†, K.A. Welsh-Bohmer8,9, J. O’Neil10,†, R. Walter11,†, S. Maresca12, M. Culp13, R. Alexander13,†, A.M. Saunders7,†, D.K. Burns7,†, C. Chiang7,†
1. Department of Psychiatry and the Behavioral Sciences, Keck School of Medicine, University of Southern California, Los Angeles, CA, USA; 2. Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago, IL, USA; 3. Department of Neurology, Indiana University School of Medicine, Indianapolis, IN, USA;
4. Department of Psychiatry and Behavioral Sciences, University of Washington, Seattle, WA, USA; 5. Department of Psychiatry, Alzheimer Disease Research Center, Icahn School of Medicine at Mount Sinai, New York, NY, USA; James J. Peters Veterans Affairs Medical Center, Bronx, NY, USA; 6. Cognitive Neuroscience Division, Department of Neurology and the Taub Institute, Vagelos College of Physicians and Surgeons, Columbia University, New York, NY, USA; 7. Zinfandel Pharmaceuticals, Inc., Durham, NC, USA; 8. Duke University Bryan Alzheimer’s Disease Research Center, Durham, NC, USA; 9. Department of Psychiatry, Duke University Medical Center, Durham, NC, USA; 10. Takeda Development Center Americas, Inc., Deerfield, IL, USA; 11. PRA Health Science, Deerfield, IL, USA; 12. IQVIA, Inc., Durham, NC, USA; 13. Takeda Development Center Americas, Inc., Cambridge, MA, USA; † Affiliation at the time of the study.
Corresponding Author: Lon S. Schneider, Keck School of Medicine of USC, 1540 Alcazar St, CHP216, Los Angeles CA, 90033, USA, Phone no: +1 323 442 7600, email: lschneid@usc.edu
J Prev Alz Dis 2022;4(9):625-634
Published online September 2, 2022, http://dx.doi.org/10.14283/jpad.2022.72
Abstract
Background: The onset of mild cognitive impairment (MCI) is an essential outcome in Alzheimer’s disease (AD) prevention trials and a compelling milestone for clinically meaningful change. Determining MCI, however, may be variable and subject to disagreement. Adjudication procedures may improve the reliability of these determinations. We report the performance of an adjudication committee for an AD prevention trial.
Methods: The TOMMORROW prevention trial selected cognitively normal participants at increased genetic risk for AD and randomized them to low-dose pioglitazone or placebo treatment. When adjudication criteria were triggered, a participant’s clinical information was randomly assigned to a three-member panel of a six-member independent adjudication committee. Determination of whether or not a participant reached MCI due to AD or AD dementia proceeded through up to three review stages – independent review, collaborative review, and full committee review – requiring a unanimous decision and ratification by the chair.
Results: Of 3494 participants randomized, the committee adjudicated on 648 cases from 386 participants, resulting in 96 primary endpoint events. Most participants had cases that were adjudicated once (n = 235, 60.9%); the rest had cases that were adjudicated multiple times. Cases were evenly distributed among the eight possible three-member panels. Most adjudicated cases (485/648, 74.8%) were decided within the independent review (stage 1); 14.0% required broader collaborative review (stage 2), and 11.1% needed full committee discussion (stage 3). The primary endpoint event decision rate was 39/485 (8.0%) for stage 1, 29/91 (31.9%) for stage 2, and 28/72 (38.9%) for stage 3. Agreement between the primary event outcomes supported by investigators’ clinical diagnoses and the decisions of the adjudication committee increased from 50% to approximately 93% (after around 100 cases) before settling at 80–90% for the remainder of the study.
Conclusions: The adjudication process was designed to provide independent, consistent determinations of the trial endpoints. These outcomes demonstrated the extent of uncertainty among trial investigators and agreement between adjudicators when the transition to MCI due to AD was prospectively assessed. These methods may inform clinical endpoint determination in future AD secondary prevention studies. Reliable, accurate assessment of clinical events is critical for prevention trials and may mean the difference between success and failure.
Key words: Mild cognitive impairment, adjudication, methodology, randomized clinical trial, delay of onset of MCI due to AD, pioglitazone.
Abbreviations: AD: Alzheimer’s disease; CAF: Clinical Assessment Form; CDR: Clinical Dementia Rating; CIAC: Cognitive Impairment Adjudication Committee; CMFV: comprehensive medical follow-up visit; MCI: mild cognitive impairment; NIA/AA: National Institute on Aging and Alzheimer’s Association.
Background
The Alzheimer’s disease (AD) therapeutic research paradigm shift from symptomatic treatment to interventions that delay or prevent clinical expression brings several new challenges for clinical trials, including recruitment, retention, endpoint determination, and whether trial outcomes are clinically meaningful (1). The assessment of cognitive decline and clinical status is required to determine the clinical impact of treatment, and it is essential to independently adjudicate endpoints to minimize the risk for bias across sites. There are several examples of endpoint adjudication in clinical trials in various therapeutic areas (2, 3), including AD (4-9) .
The TOMMORROW clinical trial (ClinicalTrials.gov NCT01931566) provides an example of a robust adjudication protocol for the determination of a primary endpoint event, namely, mild cognitive impairment (MCI) due to AD (10). It was a phase 3, multicenter, randomized, double-blind, placebo-controlled, parallel-group trial designed to: (1) qualify a biomarker risk assignment algorithm (11, 12) to determine near-term risk for MCI due to AD; and (2) evaluate the efficacy of a low dose of pioglitazone, 0.8 mg sustained release, to delay the onset of MCI due to AD in cognitively unimpaired participants assigned as high risk by the biomarker algorithm.
The adjudication process was noteworthy because, for the first time, the National Institute for Aging and Alzheimer’s Association (NIA/AA) core clinical criteria for MCI due to AD were used to define the transition of an unimpaired participant to the earliest defined stage of the AD clinical continuum as an endpoint of a registration study. This paper describes how the adjudication was conducted, examines its outcomes, and summarizes considerations that may be useful to guide future trials.
Methods
The clinical trial design has been described previously (13). Primary endpoints were the onset of MCI due to AD (10) or AD dementia (14) as adjudicated by a committee. Study visits were conducted every 6 months over a period of approximately 4 years, with anticipated study completion when 202 events would be observed in the efficacy population.
Enrollment began 1 August 2013 and was completed 21 December 2015; in total, 57 clinical sites were included from the USA, the UK, Australia, Switzerland, and Germany. While the trial originally was designed to enroll approximately 5400 participants to accrue 410 endpoint events, the protocol was modified, and this increased the expected effect size from 30% to 40%, while the number of enrolled participants was reduced to 3494. The study was terminated early by the sponsor in January 2018 for not meeting the futility analysis criteria. The trial results are reported elsewhere (15).
Clinical assessments
The two types of study visit after baseline were: (1) an in-clinic visit scheduled at 6‑month intervals and (2) a comprehensive medical follow-up visit (CMFV) that occurred usually within 30 days after a participant met protocol-specified trigger criteria at the regular 6-month visit. The assessments collected at these visits are listed in Fig. 1. The 6-month visits were intended to evaluate safety and to assess clinical and neuropsychological function, using information obtained from the participant and their study partner. The CMFV required additional assessments to aid the site investigator’s clinical diagnosis when a 6-monthly interval visit suggested a change in a participant’s cognitive status. After each CMFV, the investigator assigned a provisional clinical diagnosis to the participant’s case from the following six categories: cognitively normal; reversible cognitive impairment (e.g., from a medical condition or medication); irreversible cognitive impairment (e.g., from a medical condition such as a stroke or tumor); MCI due to AD; AD dementia; or non-AD dementia. Following a CMFV, participants continued regular 6-monthly visits unless a site investigator decided to withdraw the participant from the trial or an adjudication decision determined that the participant met the criteria for a primary endpoint.

Figure 1. Study assessments at 6-month interval visits and CMFVs
Regular study visits were scheduled for participants and their study partners every 6 months after the randomization visit. If any CMFV trigger criterion was met, participants and partners were required to attend a medical follow-up within 30 days of the visit at which the trigger criterion was met. The CDR and neuropsychological battery were both centrally reviewed, providing feedback to the sites to provide consistency between sites. *Post-baseline assessments conducted yearly (every 12 months). †This battery is a novel combination of widely used tests: California Verbal Learning Test II, Brief Visuospatial Memory Test-Revised, Trail Making Test (Parts A/B), Clock-Drawing Test, Controlled Oral Word Association Test, Animal Fluency Test, Digit Span (forward/backward), and Multilingual Naming Test. AD, Alzheimer’s disease; ADCS, Alzheimer’s Disease Cooperative Study; ADL-PI, Activities of Daily Living – Prevention Instrument; CDR, Clinical Dementia Rating; CGIC-MCI, Clinical Global Impression of Change – Mild Cognitive Impairment; CMFV, comprehensive medical follow-up visit; C-SSRS, Columbia–Suicide Severity Rating Scale; ECG, electrocardiogram; GDS, Geriatric Depression Scale; IQCODE, Informant Questionnaire on Cognitive Decline in the Elderly; MCI, mild cognitive impairment; MMSE, Mini-Mental State Examination; MRI, magnetic resonance imaging; NPI-Q, Neuropsychiatric Inventory Questionnaire; SD, standard deviation
Initiation of case adjudication
Fig. 2 summarizes the protocol-defined criteria that would initiate adjudication of a case. The first criterion was a determination that a participant met CMFV trigger criteria, that is, suspicion of cognitive decline, at two consecutive study visits. The second criterion required an MCI due to AD diagnosis by an investigator after any CMFV. In these situations, the participant continues in the trial and returns for the next 6-month interval visit. This visit would require a CMFV regardless of whether the trigger criteria were met so that the adjudication review would have data from two consecutive visits upon which to base a decision regarding the primary endpoint event. The third criterion was an investigator’s diagnosis of AD dementia, non-AD dementia, or irreversible cognitive impairment. Each of these diagnoses presented the possibility that a participant had declined to AD dementia, which was also a primary endpoint event. Therefore, these cases were assigned for adjudication after the CMFV when the diagnosis was made.
Cases meeting these adjudication initiation criteria were prepared for adjudication review by a vendor company (IQVIA, Research Triangle Park, NC, USA), using an electronic platform (Merge eClinical Operating System) to create a dossier that included clinical profile information (e.g., demographics, cognitive battery profile sheets, and noncognitive assessment summaries), investigator clinical narratives, visit source worksheets, Clinical Dementia Rating (CDR) scale worksheets, Clinical Global Impression of Change source documents, laboratory reports for values reported as adverse events, and brain imaging scans and reports (if applicable). To provide robust and consistent case details, the study team implemented guidance for site investigators regarding their clinical narratives by standardizing a Clinical Assessment Form (CAF) to be included with each dossier. The CAF narrative was required for any clinical diagnosis other than “cognitively normal” and provided a description of disease onset (e.g., “abrupt” or “insidious”) and disease course (e.g., slowly progressive versus stepwise), a neurocognitive profile (neuropsychologist report), a summary of other clinical data, and comments on any complicating medical factors.
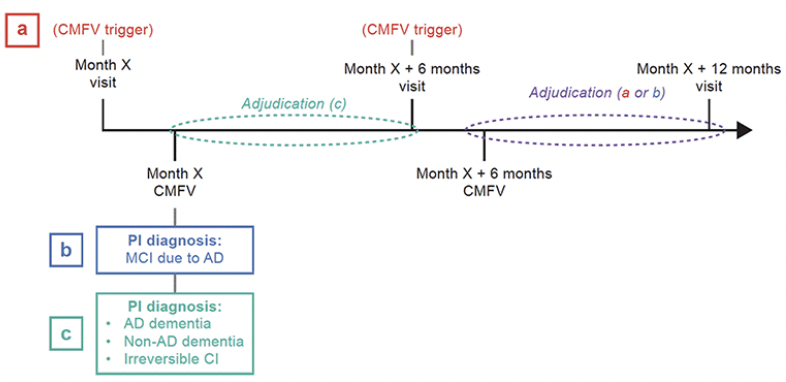
Figure 2. Adjudication initiation criteria
Three criteria determined the need for case adjudication: (a) case is forwarded after two consecutive CMFVs if CMFV trigger criteria are met at two consecutive 6-month interval study visits; (b) case is forwarded after two consecutive CMFVs if the investigator’s first clinical diagnosis is “MCI due to AD”, here, the second CMFV is mandatory and not conditioned on triggering; and (c) case is forwarded after a single CMFV if the investigator’s clinical diagnosis is “AD dementia”, “non-AD dementia”, or “irreversible CI”. Dotted ovals indicate the relative time frames during which case adjudication would occur: in a or b, adjudication takes place after the second of two consecutive CMFVs, while in c, adjudication takes place after the CMFV when the investigator’s diagnosis is AD dementia, non‑AD dementia, or irreversible CI. AD, Alzheimer’s disease; CI, cognitive impairment; CMFV, comprehensive medical follow-up visit; MCI, mild cognitive impairment; PI, principal investigator
CIAC composition and remit
The Cognitive Impairment Adjudication Committee (CIAC) comprised seven independent experts. One served as chair and had responsibility for committee oversight and ensuring endpoint event harmonization, that is, the appropriate and consistent application of MCI due to AD and AD dementia diagnostic criteria in endpoint event decisions across sites. Six served as reviewing members, with two each having expertise in neurology, psychiatry, and clinical neuropsychology. Members did not otherwise participate in the conduct of the trial.
A stratified random assignment method was used to assign each dossier to a three‑member review panel, with one expert each in neurology, psychiatry, and neuropsychology (Fig. 3). Eight different combinations of reviewing members were randomly assigned dossiers in this manner.
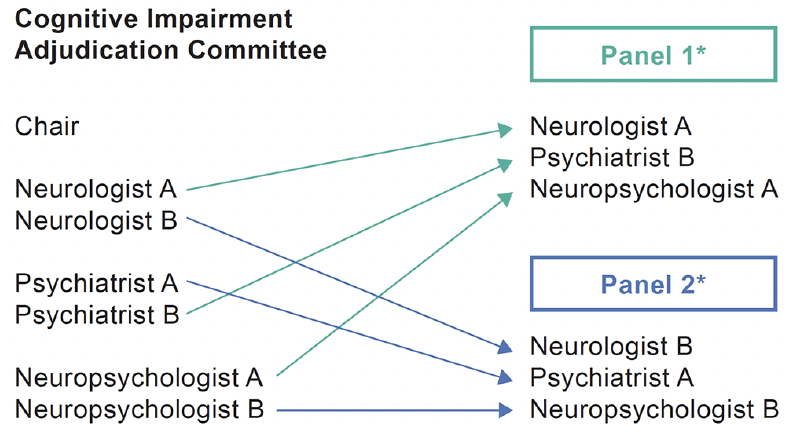
Figure 3. Cognitive Impairment Adjudication Committee
The committee responsible for the adjudication of primary endpoint events was comprised of one chair and six reviewing members. To review a case, the adjudication vendor assigned a dossier to a randomly selected panel of three reviewers, each representing a different discipline. *Panels are examples of eight possible combinations of Cognitive Impairment Adjudication Committee reviewers
The committee’s remit was to review data within the Merge web-based study portal for participants suspected of cognitive decline and to provide a primary endpoint event outcome decision on whether or not the participants met criteria for MCI due to AD or AD dementia. According to the study protocol, “MCI due to AD” was recorded when a participant met core clinical criteria for MCI due to AD at two consecutive study visits and CMFVs, and “AD dementia” was recorded when a participant met the NIA/AA criteria at any single study visit and CMFV (14).
Adjudication procedures
The method for adjudication adapted from Gabel et al. (2010) (16) was: (1) the presentation of a uniform set of information to the review panel; (2) an initial independent decision from each reviewer, which is recorded and subsequently shared with the other reviewers; (3) discussion by the reviewers of their opinions; and (4) a final group decision requiring consensus. Adjudicators were blinded to the risk and treatment strata of participants whose cases were to be adjudicated (16).
After a case was assigned, the process could progress through up to three stages; the required unanimous agreement could occur at any of these stages (Fig. 4). Initially (stage 1), the three members independently reviewed each dossier and logged their event decision within 14 days. If the three reviewers reached the same decision, the dossier and reviews were sent to the chair. If the chair ratified the decision, the decision was shared with the investigator. If the chair did not agree with the decision, the dossier would be reviewed by all committee members at a monthly teleconference. Stage 2 of the process was initiated if agreement was not reached after the independent review; the three reviewers shared each other’s comments and decisions, and each had an additional 7 days to reevaluate their own decision. Agreement at the end of this stage resulted in the same procedures that were followed at stage 1, but if unanimity was still not reached, or if the chair did not ratify the decision, the dossier was reviewed at the next monthly teleconference (stage 3) by all committee members. This meeting could not adjourn until unanimity was reached on the endpoint decision. If agreement could still not be reached, the chair was empowered to cast the deciding vote.
While the adjudication committee’s endpoint decision determined the participant’s disposition, the site investigator’s clinical diagnosis was expected to guide decisions regarding clinical management because a site investigator is ultimately responsible for a research participant’s care and well-being. During the adjudication process, committee members could make requests for additional information, which were conveyed through the vendor.
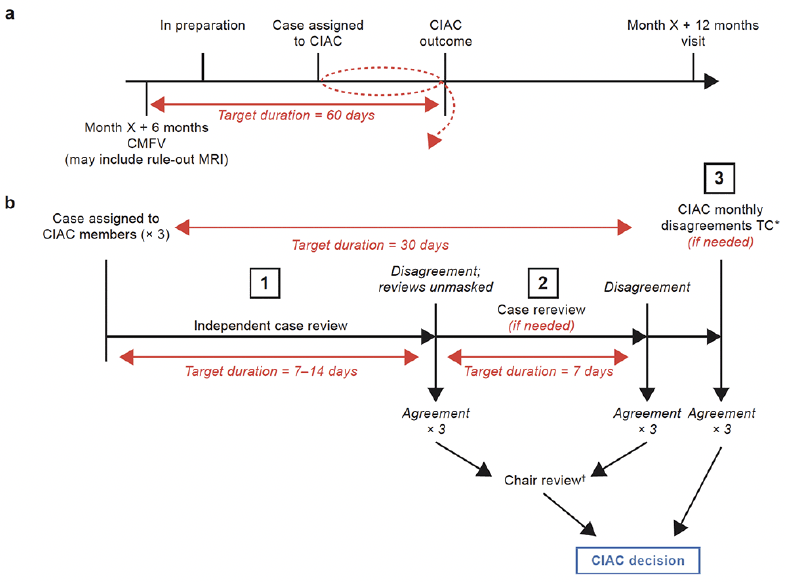
Figure 4. Adjudication process. Process flow and timing for adjudications
a After a CMFV when an adjudication initiation criterion is met, the participant’s dossier is assigned to a randomly comprised review group. b In stage 1, a reviewing member has 1–2 weeks to determine if an endpoint event occurred. If all three reviewers independently agree, then the chair may ratify the decision or assign the dossier for additional discussion at a meeting attended by all CIAC members. If all three reviewers do not agree after independent review, the process progresses to stage 2, at which the adjudicators are able to access each other’s comments and given 1 week to reevaluate their decision. Continued disagreement results in dossier review at stage 3, a monthly teleconference attended by all CIAC members. The target maximum duration of the process is 30 days. *Attended by all six members and the chair. †If the chair does not ratify the decision of the reviewers, the case will be assigned to the monthly disagreements TC. CIAC, Cognitive Impairment Adjudication Committee; CMFV, comprehensive medical follow-up visit; MRI, magnetic resonance imaging; TC, teleconference
Results
Of the 57 clinical sites, 51 (89.5%) had at least one participant meet CMFV trigger criteria, 47 sites (82.5%) had at least one participant adjudicated, and 28 (49.1%) had at least one meet endpoint event criteria. With greater than 8400 patient-years’ exposure to study medication, 974/3494 (27.9%) participants met trigger criteria for a CMFV at least once during the trial; 472/3494 (13.5%) met the criteria at least twice; and 100/3494 (2.9%) met the criteria four or more times. The most common reason for a CMFV was that a participant met thresholds for possible impairment on the neuropsychological tests (93%).
In total, 648 dossiers were adjudicated, representing 386 individuals. Most of these individuals had cases that were adjudicated only once (235, 60.9%), while the remaining 151 had cases that were adjudicated multiple times. Table 1 provides a demographic summary of these participants categorized by their final adjudication. In total, 96 participants were adjudicated to have met the endpoint event criteria for either MCI due to AD (n = 92) or AD dementia (n = 4), while 290 were adjudicated as not having met these criteria. No clear demographic differences were apparent between participants meeting MCI criteria and those considered to have neither MCI nor AD. There was a somewhat greater proportion of APOE ε4 genotype carriers among the participants adjudicated as having MCI due to AD than those who were not (61% vs 54%). All four of the participants adjudicated as having met the criteria for AD dementia were APOE ε3/ε4 carriers and somewhat older than the other participants.
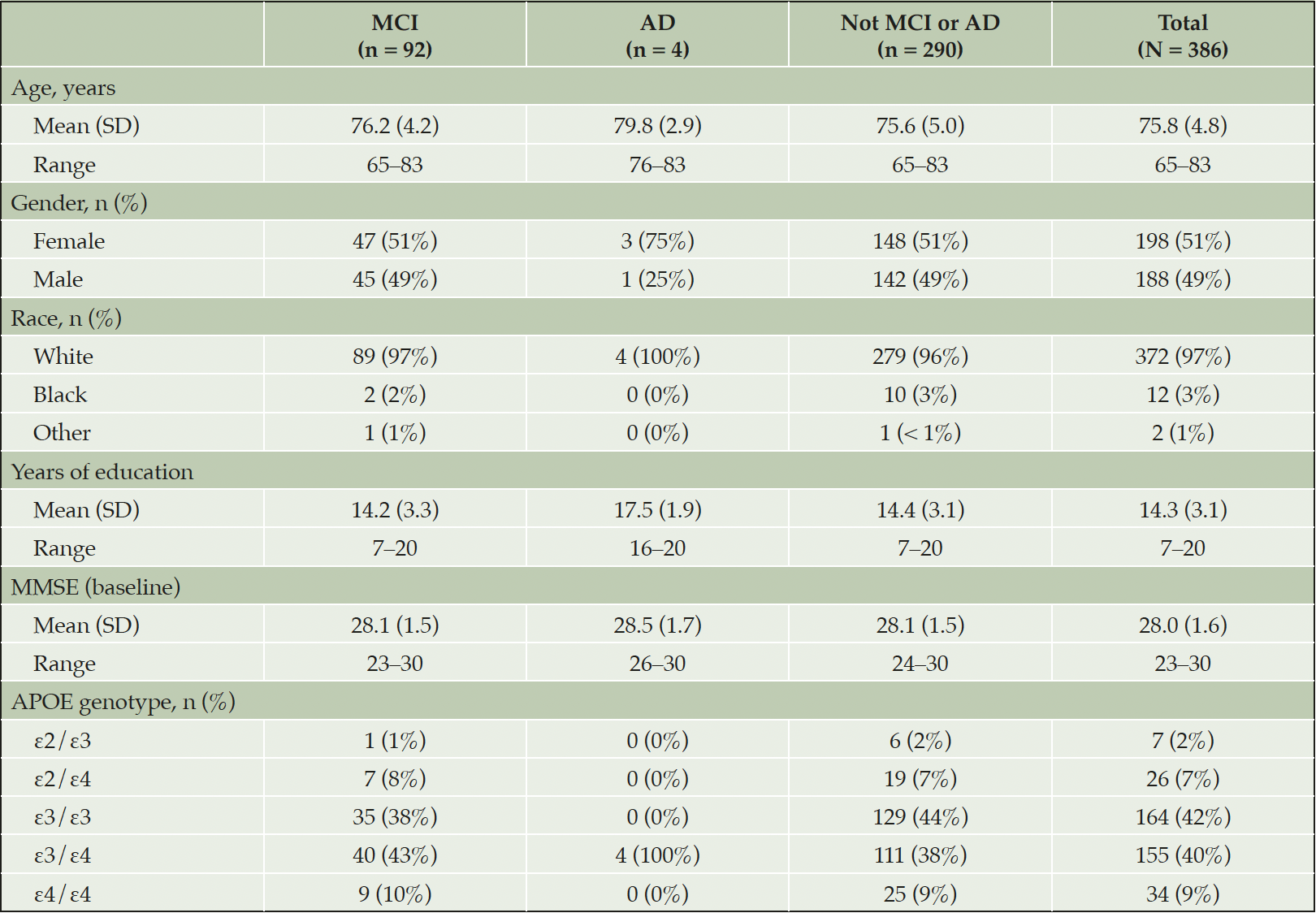
Table 1. Demographic data for participants whose dossiers were adjudicated
Participants are categorized by the final event decision reached by the adjudication committee; for those whose dossiers were adjudicated more than once, the last adjudication decision was used to determine their status for this table. AD, Alzheimer’s disease; MCI, mild cognitive impairment; MMSE, Mini-Mental State Examination; SD, standard deviation
Based on the adjudication initiation criteria in Fig. 2, in total, 63.3% (410/648) of participants met criterion “a” (two consecutive CMFV triggers) and had dossiers forwarded for adjudication; 29.5% (191/648) met criterion “b” (investigator provisional diagnosis of MCI due to AD); and 7.3% (47/648) met criterion “c” (investigator provisional diagnosis of AD or non-AD dementia or irreversible cognitive impairment). Dossiers were evenly distributed among the eight review groups, with no single group reviewing fewer than 11.1% (72/648, Group F) or greater than 14.5% (94/648, Group B) of dossiers (additional file 1, supplementary information). The decision rates that resulted in the 92 MCI endpoint events ranged from 9.2% (7 of 76 assigned cases; Group C) to 23.6% (17 of 72 assigned cases; Group F).
Breaking down the endpoint event decisions by the stage of the review when the decisions were made (Fig. 5), most adjudicated cases (485/648, 74.8%) were decided within stage 1, the independent review; 14.0% (91/648) were adjudicated in stage 2, and 11.1% (72/648) in stage 3. Stage 1 had the lowest rate of endpoint event decisions (39/485, 8.0%), stage 2 had 29/91 (31.9%), while stage 3, full committee review, had the highest rate of endpoint event decisions (28/72, 38.9%). After 3‒4 months of reviewing cases, the level of agreement across adjudicators stabilized in terms of endpoint event decisions (data not shown).
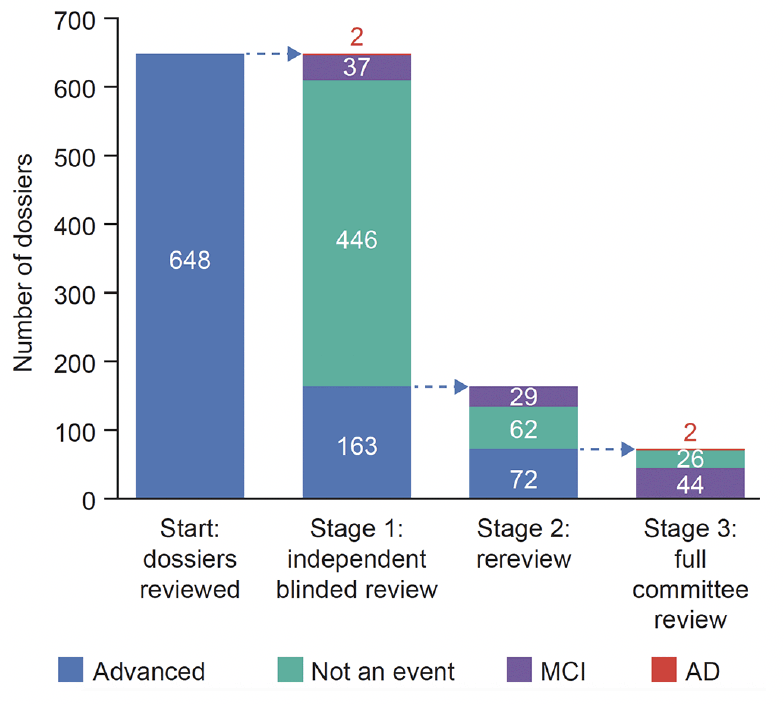
Figure 5. Primary endpoint event decisions – breakdown by review stage
This figure summarizes primary endpoint event decisions throughout the three stages of the review process – the independent review (stage 1), and, when necessary, the case rereview (stage 2) and the full committee review teleconference (stage 3). AD, Alzheimer’s disease; MCI, mild cognitive impairment
The accumulation of primary endpoint events began slowly during the recruitment phase, increased during the double-blind treatment phase, and continued after study termination until a final count of 96 events was reached (Fig. 6A). Events continued to increase after the trial was terminated and medication treatment was ended because participants who had triggered and attended a CMFV before the decision was made to end the trial were required to complete their next scheduled study visit and, if indicated, another CMFV, before ending their participation. This was done to complete the assessment of whether these participants reached an endpoint. Participants meeting the adjudication initiation criteria had their dossiers forwarded to the committee for endpoint event decision, until no more participants remained in the study.
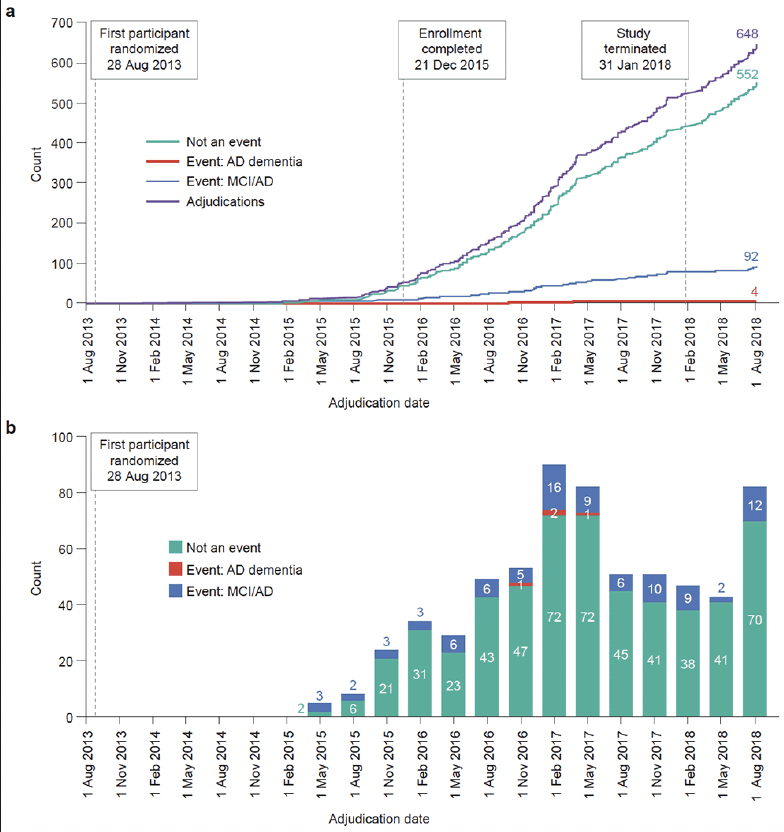
Figure 6. Primary endpoint event accumulation over time
a The number of enrolled participants at each timepoint (“enrolled count”) and the number of adjudicated primary endpoint events (“event count”). Dashed lines indicate the date that the first participant was randomized (28 August 2013), enrollment completion date (21 December 2015) and study termination date (31 January 2018). b The number of cases reviewed by the adjudication committee and their endpoint event outcomes from randomization to the end of the study. The dashed line indicates the date that the first participant was randomized (28 August 2013). AD, Alzheimer’s disease; MCI/AD, mild cognitive impairment due to Alzheimer’s disease
As expected, the cases reviewed increased over time as more participants were enrolled and followed as the trial progressed (Fig. 6B). The numbers of reviews increased and leveled off 3 years after the start of recruitment and 1 year after the last participant was randomized. For comparison, the cumulative count over time of cases reviewed, subsequently rereviewed, and then reviewed by full committee is displayed in additional file 2 (supplementary information).
A participant’s dossier included two clinical diagnoses from the investigator that resulted from two consecutive medical follow-up visits. The adjudication committee determined whether a participant met MCI due to AD criteria at both visits (or AD dementia at just one visit). Cases that were sent specifically for adjudication of possible AD dementia only needed data from a single 6-month visit and CMFV. Table 2 summarizes the combinations of investigator diagnoses from the different adjudication scenarios (described previously in Fig. 2) and their resulting adjudicated outcomes. While investigators did not offer endpoint-event opinions to the committee, their diagnoses either supported or did not support the protocol-specified criteria for an endpoint event. The table also shows how the committee’s event decisions either agreed or disagreed with that support. In the majority of the 648 adjudicated cases (60.3%, 391/648), when the adjudicated case included two diagnoses (adjudication scenarios “a” and “b” in Fig. 2), the diagnoses were either both cognitively normal (n = 275) or both MCI due to AD (n = 116). Almost half of the endpoint events (47/96, 49.0%) were derived from dossiers in which the investigator’s two diagnoses were both MCI due to AD; however, this represents only a 40.5% rate (47/116 cases) when the committee agreed with the investigators that MCI criteria were met at both visits. Most of the remainder of the event total came from situations when the case diagnoses did not support meeting the endpoint event criteria, but the committee disagreed (41/410 cases, 10%).
Agreements between outcomes supported by investigators’ diagnoses and the event decisions of the committee trended differently over time. For all 648 cases, the agreement rate rose from 50% to approximately 93% (after about 100 cases) before settling at between 80% and 90% for the rest of the study (Fig. 7A). For the 116 cases when investigator diagnoses were MCI due to AD at two consecutive CMFVs, the agreement rate fell from 100% to about 50% after half of these cases had been received, then decreased to settle at 40.5% by the end of the study (Fig. 7B).
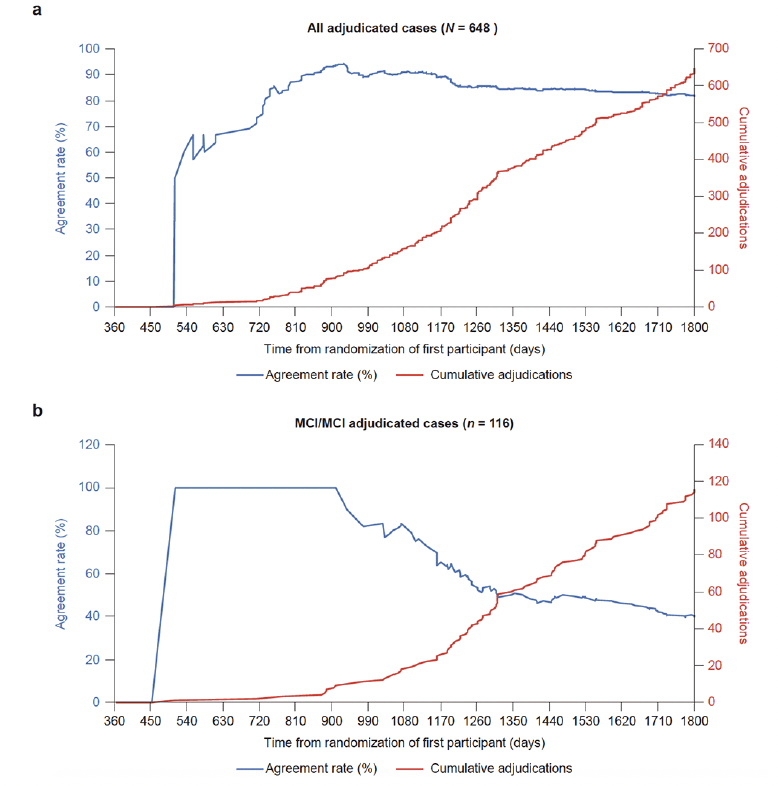
Figure 7. Agreement rates over time for CIAC event outcomes with outcomes supported by investigator diagnoses
a The blue line shows the rate at which adjudication committee endpoint decision outcomes agreed with the outcome by the site investigators’ clinical diagnoses for all 648 adjudicated cases over the course of the trial. The gray dotted line shows the accumulation of cases. b The blue line shows the rate at which adjudication committee endpoint decision outcomes agreed with the MCI due to AD outcome by the site investigators’ clinical diagnoses in 116 adjudicated cases over the course of the trial. The gray dotted line shows the accumulation of cases over the course of the trial. AD, Alzheimer’s disease; CIAC, Cognitive Impairment Adjudication Committee; MCI, mild cognitive impairment
We calculated a Cohen’s kappa for the agreement between the site investigator’s second CMFV after triggering and the adjudication committee’s diagnosis (data in Table 2). The agreement for an MCI due to AD diagnosis was 62.8% with a Cohen’s kappa = 0.327, indicating fair agreement. As there were only 4 AD dementia diagnoses in question, we also calculated agreement between the investigator’s diagnosis of MCI due to AD or AD and the adjudication committee’s diagnosis. Here, the agreement for an MCI due to AD or AD dementia diagnoses was 76.9% with a Cohen’s kappa = 0.539, indicating moderate agreement
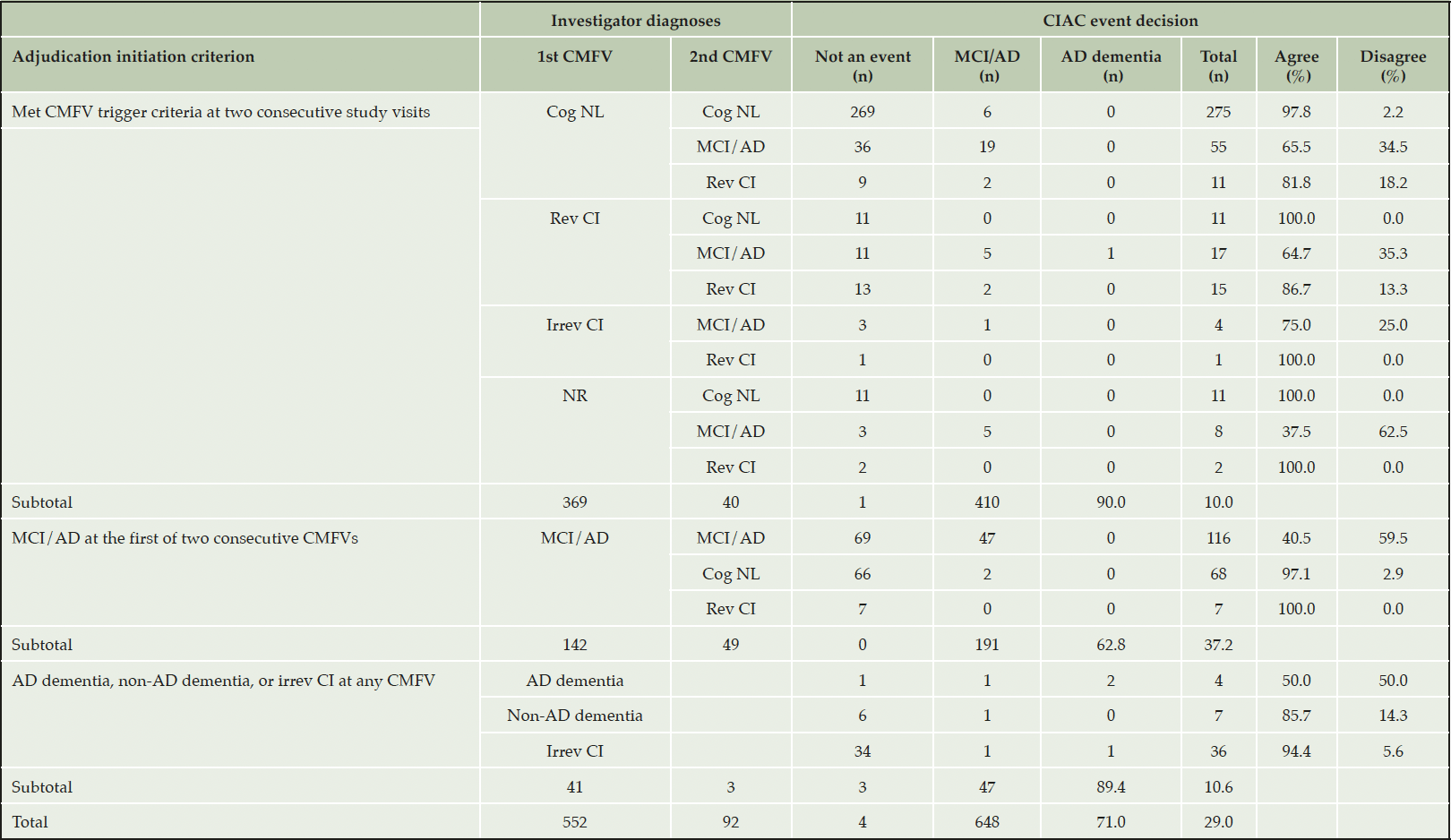
Table 2. Investigator clinical diagnoses and CIAC primary endpoint event decisions
This table shows the CIAC’s endpoint decisions as a function of the site investigator’s diagnoses for each reviewed case. CIAC, Cognitive Impairment Adjudication Committee; CMFV, comprehensive medical follow-up visit; cog NL, cognitively normal; irrev CI, irreversible cognitive impairment; MCI/AD, mild cognitive impairment due to Alzheimer’s disease; NR, investigator’s diagnosis not recorded; rev CI, reversible cognitive impairment
Discussion
The TOMMORROW trial was one of the first phase 3 trials to use onset of MCI due to AD as the primary endpoint (13). While changes in neuropathologic biomarkers are becoming essential components of long-term AD trials and may be informative surrogates for disease progression, the central challenge remains the accurate determination of clinical illness progression across the AD continuum. From a participant, caregiver, regulatory, or payer perspective, it is the most meaningful milestone that can be ascertained for a prevention or disease-progression trial. New information gleaned from using a biomarker framework for diagnosis and progression such as the NIA/AA “A/T/N” framework (17) or the US Food and Drug Administration draft guidance for early stage AD (18) does not diminish the importance of a clinical- diagnosis-based endpoint determination. Although the TOMMORROW study was truncated for futility (15), its experience in the adjudication of MCI due to AD as the primary endpoint event is instructive for future research efforts.
Like the triggering criteria, the criteria for initiating adjudication of a case were centered around neuropsychologically defined thresholds to facilitate reliable identification of the earliest transition points from cognitively unimpaired to MCI due to AD. While it was not expected that every triggered case would result in an MCI diagnosis by the investigator, casting a wider net increased the likelihood that MCI diagnoses would not be missed. Triggers were set to enable a review of participants who had two consecutive visits at which they triggered the criteria regardless of any diagnosis (or not) given by the site investigator. CIAC metrics revealed that while most adjudicated cases were decided within the initial independent review, over 90% were judged not to have met the criteria for MCI due to AD or for AD dementia (Fig. 5). However, 10% of cases adjudicated with investigator diagnoses other than MCI (at two consecutive visits) or AD dementia (at a single visit) were judged as primary endpoint events, representing close to half of the total events in the trial (41/96, 42.7%). These are events that would have been missed if the adjudications had been limited only to cases when the investigator felt an endpoint had been reached.
The majority of endpoint event determinations (57/96, 59.4%) were achieved during the rereviews (stage 2) or the full committee reviews (stage 3); and the highest rate of primary endpoint event decisions (28/72, 38.9%) occurred in the latter. The committee’s decision-making was conservative by design: when there was any doubt about the participant having met the MCI criteria, the committee made a “does not meet” decision, allowing the participant to continue in the trial for at least another 6 months. While some cases repeatedly triggered and were judged not to have met MCI criteria, it remains unknown whether they would have been given such a diagnosis had they been observed for longer durations. The implication of these results is that the committee generally found it relatively easy to determine when cases did not meet endpoint criteria and to reject those at the first stage. Determination of the MCI endpoint, however, was uncommonly a clear-cut decision easily confirmed in the first review; confirmation usually took a full review and discussion within the full committee. This result speaks to the careful consideration and conservative approach taken by the committee and to the difficulties presented by the MCI due to AD diagnostic construct itself.
The adjudication committee was comprised of clinicians in the USA. Plans to include European representation were not implemented after the trial sponsors reduced the sample size and number of sites, and when it became evident that most participants would be from English-speaking countries (98.3% from the USA, Australia, and the UK, and 1.7% from the seven German and Swiss sites). By design, every case was reviewed by three committee members each representing psychiatry, neurology, and neuropsychology to provide perspectives from different clinical viewpoints. All committee members served in their roles throughout the study, over approximately 5 years. The lack of turnover provided strong internal consistency in endpoint event decision-making throughout the trial.
Some degree of offset between site investigator diagnoses and committee endpoint decisions was expected due to the novelty of the process and the historical heterogeneity of the MCI construct. In fact, only half of the MCI (47/92, 51%) or AD (2/4, 50%) decisions affirmed an investigator’s diagnosis (Table 2). However, for non-AD dementia and irreversible CI diagnoses there was 94% agreement with investigators’ diagnoses that these were not MCI due to AD or AD dementia endpoints, thus indicating a very high level of specificity for the endpoint diagnoses. The protocol intended and allowed for both investigator and committee to have the ability to consider a participant’s full clinical presentation in evaluating MCI due to AD. The committee relied substantially on clinical notes from the site investigator and neuropsychologist, finding the additional perspectives to be helpful, particularly with respect to uncertain cases. For example, in cases when an investigator’s diagnosis of MCI due to AD was adjudicated as a primary endpoint event without a CDR score of 0.5, sites were required to capture diagnostic reasoning on the CAF and to acknowledge the CDR scores. CDR scores were also highlighted for the committee to bring attention to cases when the score may not have aligned with the operationalized criteria, and members were required to document the CDR element in their entry of adjudication decisions.
Limitations
The adjudication process had several noteworthy limitations. There was no systematic scale of behavior or psychiatric symptoms used that otherwise may have been sensitive early detection variables for MCI but are not well-established as such. Robust longitudinal data for participants with MCI are lacking that might have shown progression to AD because only 40 individuals who received a diagnosis of MCI continued in the blinded extension study. That study was also stopped early when the trial was stopped for futility.
AD neuropathologic biomarkers were not prospectively collected in the trial. However, using a neural network model to impute amyloid plaques from MRI data we estimated that 83% of participants who progressed to MCI due to AD were plaque-positive (Alexander R et al. Oral presentation, Alzheimer’s Association International Conference July 17, 2019, unpublished). Moreover, converters to MCI generally had lower overall cognitive scores at baseline. Thus, the high-risk participants who met eligibility criteria for this trial, based on APOE and TOMM40 genotypes and age, were similar to the preclinical AD participants of recent amyloid antibody trials.
Conclusions
Although adjudication for a clinical outcome such as probable AD dementia or MCI is commonly done in large-scale AD clinical trials, the adjudication process in this trial and its metrics provide unique insight into the way this can work in future large-scale trials when the therapeutic goal is to delay clinical progression or the onset of early-stage AD.
Clinical Site Investigators: Please refer to additional file 3 (supplementary information).
Ethics approval and consent to participate: Not applicable.
Consent for publication: Not applicable.
Availability of data and materials: The datasets, including the redacted study protocol, redacted statistical analysis plan, and individual participants data supporting the results reported in this article, will be made available within 3 months from initial request, to researchers who provide a methodologically sound proposal. The data will be provided after its de-identification, in compliance with applicable privacy laws, data protection and requirements for consent and anonymization. Research proposal applications can be made to www.Vivli.org where they will be reviewed and approved by an independent review panel. After a signed data access agreement has been received, the study’s protocol, statistical analysis plan, blank case report forms, clinical study report, de-identified individual patient data, and a data dictionary defining each field in the dataset will be made available at www.Vivli.org.
Competing interests: LSS received personal fees from Takeda Pharmaceuticals during the conduct of the study and served as Chair of the TOMMORROW study Cognitive Impairment Adjudication Committee. DAB received personal fees from Takeda Pharmaceuticals during the conduct of the study. Outside of the submitted work, LSS reports grants and personal fees from Eli Lilly, Merck, Roche/Genentech; personal fees from Avraham Ltd, Boehringer Ingelheim, Neurim Ltd, Neuronix Ltd, Cognition, Eisai, Takeda, vTv, Abbott, and Samus; grants from Biogen, Novartis, Biohaven, and Washington University/ NIA DIAN-TU. CC, DKB, SH, and AMS were full-time employees of Zinfandel Pharmaceuticals during study conduct. In addition, AMS has a patent TOMM40 Biomarker Algorithm issued, a patent Low Dose Pioglitazone in MCI issued, and a patent Extended Release Formulation Low Dose Pioglitazone issued. RCA, MC, JO, and RW were full-time employees of Takeda Pharmaceuticals during study conduct. KAW-B received funding from Takeda Pharmaceuticals for her work on the project as part of the Neuropsychology Lead Office at Duke University. Outside the submitted work, KAW-B reports personal fees from Biogen and a grant from VeraSci. MWL received consulting fees from Zinfandel Pharmaceuticals.
Funding: This study was funded by Takeda Pharmaceutical Company and Zinfandel Pharmaceuticals. Takeda Pharmaceutical Company provided funding to Oxford PharmaGenesis for editorial assistance in formatting, proofreading, copy editing, and fact checking.
Authors’ contributions: LSS, DAB, MRF, ERP, MAR, MS, YS served on the adjudication committee. CC, JO, RW, MC, SH, KW-B, SM, DKB and LSS participated in study concept and design. All authors participated in study execution. LSS, CC, SH, SM, DKB participated in data analyses. LSS, CC, SH and DKB prepared the first draft of the manuscript. All authors critically reviewed the manuscript, participated in revisions and approved the submitted version.
Acknowledgements: The authors would like to gratefully acknowledge the study participants and their project partners and the TOMMORROW site investigators, neuropsychologists, and site staff for their efforts and commitment to this important study. We also gratefully acknowledge the work of Mary Jenkins, Jeannetta Crutchfield, Sylvia Maresca, and the Clinical Event Validation and Adjudication (CEVA) team at our vendor partner IQVIA for management of the operational components of the adjudication process.
Trial registration: NCT01931566.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. Aisen P, Touchon J, Amariglio R, Andrieu S, Bateman R, Breitner J, et al. EU/US/CTAD task force: lessons learned from recent and current Alzheimer’s prevention trials. J Prev Alzheimers Dis. 2017;4(2):116–24.
2. Held C, White HD, Stewart RAH, Davies R, Sampson S, Chiswell K, et al. Characterization of cardiovascular clinical events and impact of event adjudication on the treatment effect of darapladib versus placebo in patients with stable coronary heart disease: Insights from the STABILITY trial. Am Heart J. 2019;208:65–73.
3. Farrant M, Easton JD, Adelman EE, Cucchiara BL, Barsan WG, Tillman HJ, et al. Assessment of the end point adjudication process on the results of the platelet-oriented inhibition in new TIA and minor ischemic stroke (POINT) trial: a secondary analysis. JAMA Netw Open. 2019;2(9):e1910769.
4. Shumaker SA, Legault C, Rapp SR, Thal L, Wallace RB, Ockene JK, et al. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA. 2003;289(20):2651–62.
5. Yaffe K, Krueger K, Cummings SR, Blackwell T, Henderson VW, Sarkar S, et al. Effect of raloxifene on prevention of dementia and cognitive impairment in older women: the Multiple Outcomes of Raloxifene Evaluation (MORE) randomized trial. Am J Psychiatry. 2005;162(4):683–90.
6. DeKosky ST, Fitzpatrick A, Ives DG, Saxton J, Williamson J, Lopez OL, et al. The Ginkgo Evaluation of Memory (GEM) study: design and baseline data of a randomized trial of Ginkgo biloba extract in prevention of dementia. Contemporary clinical trials. 2006;27(3):238–53.
7. Feldman HH, Ferris S, Winblad B, Sfikas N, Mancione L, He Y, et al. Effect of rivastigmine on delay to diagnosis of Alzheimer’s disease from mild cognitive impairment: the InDDEx study. Lancet Neurol. 2007;6(6):501–12.
8. Meinert CL, McCaffrey LD, and Breitner JC. Alzheimer’s disease anti-inflammatory prevention trial: design, methods, and baseline results. Alzheimers Dement. 2009;5(2):93–104.
9. Tariot PN, Lopera F, Langbaum JB, Thomas RG, Hendrix S, Schneider LS, et al. The Alzheimer’s prevention initiative autosomal-dominant Alzheimer’s disease trial: a study of crenezumab versus placebo in preclinical PSEN1 E280A mutation carriers to evaluate efficacy and safety in the treatment of autosomal-dominant Alzheimer’s disease, including a placebo-treated noncarrier cohort. Alzheimers Dement (N Y). 2018;4:150–60.
10. Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011;7(3):270–9.
11. Crenshaw DG, Gottschalk WK, Lutz MW, Grossman I, Saunders AM, Burke JR, et al. Using genetics to enable studies on the prevention of Alzheimer’s disease. Clin Pharmacol Ther. 2013;93(2):177–85.
12. Lutz MW, Sundseth SS, Burns DK, Saunders AM, Hayden KM, Burke JR, et al. A genetics-based biomarker risk algorithm for predicting risk of Alzheimer’s disease. Alzheimers Dement (N Y). 2016;2(1):30–44.
13. Burns DK, Chiang C, Welsh-Bohmer KA, Brannan SK, Culp M, O’Neil J, et al. The TOMMORROW study: design of an Alzheimer’s disease delay-of-onset clinical trial. Alzheimers Dement. 2019;5:661–70.
14. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr., Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging and the Alzheimer’s Association workgroup. Alzheimer’s Dement. 2011; 7(3): 263-9 2011.
15. Burns D, Alexander R, Welsh-Bohmer K, Culp M, Chiang C, O’Neil J, et al. Safety and efficacy of pioglitazone for delay of mild cognitive impairment in people at risk of Alzheimer’s disease (TOMMORROW): a prognostic biomarker study and a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021; 20(7): 537-47.
16. Gabel MJ, Foster NL, Heidebrink JL, Higdon R, Aizenstein HJ, Arnold SE, et al. Validation of consensus panel diagnosis in dementia. Arch Neurol. 2010;67(12):1506–12.
17. Jack CR, Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. 2016;87(5):539–47.
18. US Food and Drug Administration CfDEaR. Alzheimer’s disease: developing drugs for treatment, guidelines for industry. 2018 https://www.fda.gov/regulatory-information/search-fda-guidance-documents/alzheimers-disease-developing-drugs-treatment-guidance-industy. Accessed March 2021.