
C. Mallinckrodt1,*,**, Y. Tian1,*, P.S. Aisen2, F. Barkhof3,4, S. Cohen5, G. Dent1, O. Hansson6,7, K. Harrison1,
T. Iwatsubo8, C.J. Mummery9, K.K. Muralidharan1, I. Nestorov1, L. Nisenbaum1,**, R. Rajagovindan1,**,
C. von Hehn1,**, C.H. van Dyck10, B. Vellas11, S. Wu1, Y. Zhu1, A. Sandrock1,**, T. Chen1, S. Budd Haeberlein1
1. Biogen, Cambridge, MA, USA; 2. Alzheimer’s Therapeutic Research Institute, University of Southern California, San Diego, CA, USA; 3. Department of Radiology & Nuclear Medicine, Amsterdam UMC, Amsterdam, the Netherlands; 4. UCL Queen Square Institute of Neurology & Centre for Medical Image Computing, London, UK; 5. Toronto Memory Program, Toronto, ON, Canada; 6. Clinical Memory Research Unit, Department of Clinical Sciences Malmö, Lund University, Malmö, Sweden;
7. Memory Clinic, Skåne University Hospital, Malmö, Sweden; 8. Department of Neuropathology, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan; 9. Dementia Research Centre, Queen Square Institute of Neurology, University College London, London, UK; 10. Alzheimer’s Disease Research Unit, Yale School of Medicine, New Haven, CT, USA; 11. Toulouse Gerontopole University Hospital, Universite Paul Sabatier, INSERM U 1295, Toulouse, France; * These authors contributed equally to this work; ** Denotes an author who was an employee of Biogen at the time of this work who has since left the company.
Corresponding Author: Samantha Budd Haeberlein, 225 Binney Street, Biogen, Cambridge, Massachusetts, 617-679-3159, Samantha.BuddHaeberlein@biogen.com
J Prev Alz Dis 2023;2(10):171-177
Published online January 12, 2023, http://dx.doi.org/10.14283/jpad.2023.6
Abstract
Objectives: Efficacy and safety results from the EMERGE (NCT02484547) and ENGAGE (NCT02477800) phase 3 studies of aducanumab in early Alzheimer’s disease (AD) have been published. In EMERGE, but not in ENGAGE, high-dose aducanumab demonstrated significant treatment effects across primary and secondary endpoints. Low-dose aducanumab results were consistent across studies with non-significant differences versus placebo that were intermediate to the high-dose arm in EMERGE. The present investigation examined data from EMERGE and ENGAGE through post-hoc analyses to determine factors that contributed to discordant results between the high-dose arms of the two studies.
Design: EMERGE and ENGAGE were 2 phase 3, randomized, double-blind, placebo-controlled, parallel-group studies.
Setting: EMERGE and ENGAGE were 2 global multicenter studies involving 348 sites in 20 countries.
Participants: Participants in EMERGE and ENGAGE were aged 50 to 85 years and had mild cognitive impairment or mild AD dementia with confirmed amyloid pathology. The randomized and dosed population (all randomized patients who received at least one dose of study treatment) included 1638 patients in EMERGE and 1647 in ENGAGE.
Intervention: In EMERGE and ENGAGE, participants were randomized to receive low- or high-dose aducanumab or placebo (1:1:1) once every 4 weeks.
Measurements: In this paper, 4 areas were investigated through post-hoc analyses to understand the discordance in the high-dose arms of the EMERGE and ENGAGE studies: baseline characteristics, amyloid-related imaging abnormalities, non-normality of the data, and dosing/exposure to aducanumab.
Results: Post-hoc analyses showed that outcomes in the ENGAGE high-dose group were affected by an imbalance in a small number of patients with extremely rapid progression and by lower exposure to the target dose of 10 mg/kg. These factors were confounded and present in early enrolled patients but were not present in later-enrolled patients who were randomized to the target dosing regimen of 10 mg/kg after titration. Neither baseline characteristics nor amyloid-related imaging abnormalities contributed to the difference in results between the high-dose arms.
Conclusions: Results were consistent across studies in later enrolled patients in which the incidence of rapidly progressing patients was balanced across treatment arms.
Key words: Aducanumab, Alzheimer’s disease, amyloid beta.
Introduction
Aducanumab is a human monoclonal antibody that selectively targets aggregated forms of amyloid beta (Aβ), including soluble oligomers and insoluble fibrils (1, 2). The clinical efficacy and safety of aducanumab were assessed in patients with early Alzheimer’s disease (AD) via 2 identically designed, phase 3, randomized clinical trials, EMERGE and ENGAGE; the primary findings have been reported (3). Aducanumab was granted accelerated approval by the US Food and Drug Administration based on its ability to reduce a defining pathophysiological feature of Alzheimer’s disease, Aβ plaques (2-5).
ENGAGE failed to meet its primary and secondary endpoints, whereas EMERGE demonstrated statistically significant mean differences vs placebo in the high-dose arm on the primary and the 3 multiplicity-adjusted secondary endpoints (Table S1) (3). Results of the low-dose arms were similar in the 2 studies, with differences from placebo intermediate to the differences seen in the high-dose arm of EMERGE, but nonsignificant (3). The probability that all 4 of the clinical endpoints in the high-dose arm of EMERGE were false-positive results was extremely low given the low to moderate correlations between endpoints. However, the nonsignificant results in the high-dose arm of the identically designed ENGAGE study were also unlikely given the results of the high-dose arm in EMERGE. Based on a key protocol amendment, the target dose for aducanumab was increased part-way through the trials in apolipoprotein E (ApoE) ε4 carriers (approximately 2/3 of the patients) in the high-dose arms. To understand whether dose exposure contributed to these findings and to reconcile the observed discordance in the high-dose arms of the studies, we examined potential sources of the discordance and synthesized the totality of data from the aducanumab clinical development program. The following analyses were post-hoc in nature. To the extent possible, the hypothesis and analytical approaches were specified before analyses were conducted. Results should be interpreted in this context.
Methods
Study design and data sets
Full details of the study designs have been reported (3). Briefly, EMERGE (N=1638) and ENGAGE (N=1647) (ClinicalTrials.gov: NCT02484547 and NCT02477800, respectively) were global trials involving 348 sites in 20 countries. Patients aged 50 to 85 years who met criteria for mild cognitive impairment due to AD or mild AD dementia, with confirmed amyloid pathology, were randomized 1:1:1 to receive low-dose aducanumab, high-dose aducanumab, or placebo. The primary outcome was change from baseline to week 78 on the Clinical Dementia Rating–Sum of Boxes (CDR-SB).
The EMERGE and ENGAGE trials were terminated early for futility based on prespecified interim analyses (3). However, after termination, it was discovered that key assumptions underlying the futility determination did not hold, as discussed in Budd Haeberlein S, et al. 2022 (3): a) the assumption that the treatment effect in the 2 studies would be similar and b) the assumption that the treatment effect would not change substantially over time. Consequently, the predictions of final trial outcomes were inaccurate, and the trials should not have been terminated.
We present analyses on the final data set for these studies (3). The final data set included all randomized patients and was thus much larger than the futility data set. The futility data set was defined a priori to include the data from (approximately) the first 50% of enrolled patients. Final database lock was on November 13, 2019, for EMERGE and November 15, 2019, for ENGAGE. The final primary efficacy analysis only included efficacy data collected under double-blind conditions up to March 20, 2019—the day prior to the futility announcement, and this final primary efficacy analysis included approximately twice as many patients and increased the total number of data observation by 65.2%, compared with the futility analysis. By the time of the futility analysis, enrollment for the 2 studies had been completed and all patients had the opportunity to complete at least 6 months of treatment. Results from this larger (final) data set are the focus of this article.
Analyses
Unless otherwise stated, these post-hoc analyses were conducted using the mixed model for repeated measures (MMRM) analyses, the same method as the prespecified primary analysis in the statistical analysis plan and that described in the primary disclosure of these study results (3).
Baseline demographics/disease characteristics
Analyses (using placebo data only) were conducted by study and with studies pooled. Variables were selected by stepwise model selection based on the Bayesian information criterion from the following candidate predictors: baseline scores for CDR-SB, Mini-Mental State Examination (MMSE), Alzheimer’s Disease Assessment Scale–Cognitive Subscale, 13 items (ADAS-Cog13), Alzheimer’s Disease Cooperative Study–Activities of Daily Living Inventory, mild cognitive impairment version (ADCS-ADL-MCI), and the Repeatable Battery for the Assessment of Neuropsychological Status delayed memory index, ApoE ε4 carrier status, ApoE genotype, ApoE allele pairs, baseline AD medication use and category, baseline disease stage, time since first AD symptom, time since first AD diagnosis, age, sex, baseline weight, baseline body mass index, years of formal education, and days between baseline CDR-SB and first dose date.
Accounting for imbalance in rapidly progressing patients
Two statistical methods, robust and quantile regression, were implemented to account for the non-normality of the data arising from the small number of rapidly progressing patients. The main purpose of robust regression is to provide stable estimates of means in the presence of outliers. To achieve this stability, robust regression limits the influence of outliers through a process known as iteratively reweighted least squares. Conceptually, the iterative process detects unusual observations and gives them less weight (i.e., a smaller contribution) in estimating means (6). The specific type of robust regression used here was based on M estimation (7) as implemented in SAS PROC ROBUSTREG (8). Quantile regression is also robust to extreme values of the response variable (outliers). This method was introduced by Koenker and Bassett (1978) as an extension of traditional regression that models the relationship between covariates and the mean of the response variable Y to model quantiles of the response variable, such as the median (9). Quantile regression was implemented in the present investigation to compare medians between treatment groups using SAS PROC QUANTREG (8)
Other analyses
Other analyses were conducted as described within the results section.
Availability of data and materials
The authors and Biogen are fully supportive of data sharing. Biogen has established processes to share protocols, clinical study reports, study-level data, and de-identified patient-level data. These data and materials will be made available to qualified scientific researchers to achieve the objective(s) in their approved, methodologically sound research proposal following US and EU marketing approval of aducanumab for the treatment of AD, with no end date. Proposals should be submitted through Vivli (https://vivli.org). To gain access, data requestors will need to sign a data sharing agreement. Data are made available for 1 year on a secure platform. For general inquiries, please contact datasharing@biogen.com. Biogen’s data-sharing policies and processes are detailed on the website http://clinicalresearch.biogen.com.
Results
Understanding the discordance in the high-dose arms of EMERGE and ENGAGE
Four areas were investigated to understand the discordance in the high-dose arms of the EMERGE and ENGAGE studies: baseline characteristics, amyloid-related imaging abnormalities (ARIA), non-normality of the data, and dosing/exposure to aducanumab.
Baseline demographics/disease characteristics
For a factor to substantially contribute to the divergence in results between the high-dose arms, that factor must have an appreciable influence on outcomes and be substantially unbalanced across treatment arms and studies. Analysis of baseline covariates showed that they jointly accounted for approximately 20% of the differences in clinical decline within the placebo group (see Analyses for details), and these factors were well balanced across treatment arms and studies (3). The highest r-square value across the various data sets was 0.24. Therefore, baseline demographic and disease characteristics had minimal influence on the divergence in results between the high-dose arms.
Impact of amyloid-related imaging abnormalities
ARIA is an adverse event observed in clinical trials with Aβ-targeting monoclonal antibodies, such as aducanumab (2, 3, 5, 10, 11). The incidence, radiographic severity, and reported symptoms of ARIA were similar in EMERGE and ENGAGE (3, 11) (Table S2). Hence, ARIA did not contribute to the discordance in results between the high-dose arms.
ARIA has the potential to bias clinical assessments directly or through functional unblinding, because the management of ARIA required temporary dose suspension and additional monitoring procedures, and the incidence of ARIA events (either ARIA-E or ARIA-H) was higher on active drug (high-dose group: 42.1% in EMERGE; 40.8% in ENGAGE) than placebo (10.3% in EMERGE; 10.3% in ENGAGE) (Table S2). The potential for functional unblinding due to ARIA was mitigated by the separation of efficacy assessments from safety assessments and by adverse-event management. This separation required 3 assessors for each efficacy visit. One physician assessed adverse events and managed ARIA, if present. Two other assessors rated efficacy outcomes, with one rating the primary CDR-SB and the other rating the secondary outcomes.
To evaluate the impact of potential functional unblinding due to ARIA, results based on the primary analysis were compared with results from an otherwise identical analysis in which post-ARIA observations were removed. These 2 sets of analyses yielded similar mean differences from placebo (Table S3), suggesting that there was no bias from functional unblinding. For a more granular assessment of the potential impact of ARIA, subgroups were defined by study, dose, and ApoE ε4 status (Figure S1). Comparing the mean differences vs placebo from all data (x-axis) vs data excluding observations after ARIA onset (y-axis) showed results scattered evenly above and below the line of unity, indicating random variability and no systematic bias in the post-ARIA observations. If functional unblinding biased results, the data points would be consistently below or above the unity line.
Non-normality of the data
Standard statistical diagnostic tests of the primary analysis (MMRM) indicated that the assumption of normally distributed residuals had been violated. Clinical outcomes in EMERGE and ENGAGE were right skewed due to a small number (~1%) of rapid progressors over 78 weeks on the CDR-SB (>8 points on CDR-SB change from baseline over 78 weeks) (Figure S2). The number of rapid progressors was similar in all treatment arms of both studies, except for a higher incidence in the high-dose arm of ENGAGE (Table S4). The consequences of this non-normality were investigated in post-hoc analyses by comparing results from the prespecified MMRM primary analyses that assumed normality to those from robust regression and quantile regression.
Table 1 summarizes results from robust and quantile regression analyses, which unlike the primary MMRM analysis, did not depend on normality. In both studies, treatment effect estimates for the high-dose arm were larger in the alternative analysis than in the primary MMRM analysis. Accordingly, the 1% of patients classified as rapid progressors had an important influence on mean differences vs placebo. Differences between the balance of rapid progressors in EMERGE and ENGAGE are described in detail later in this paper.
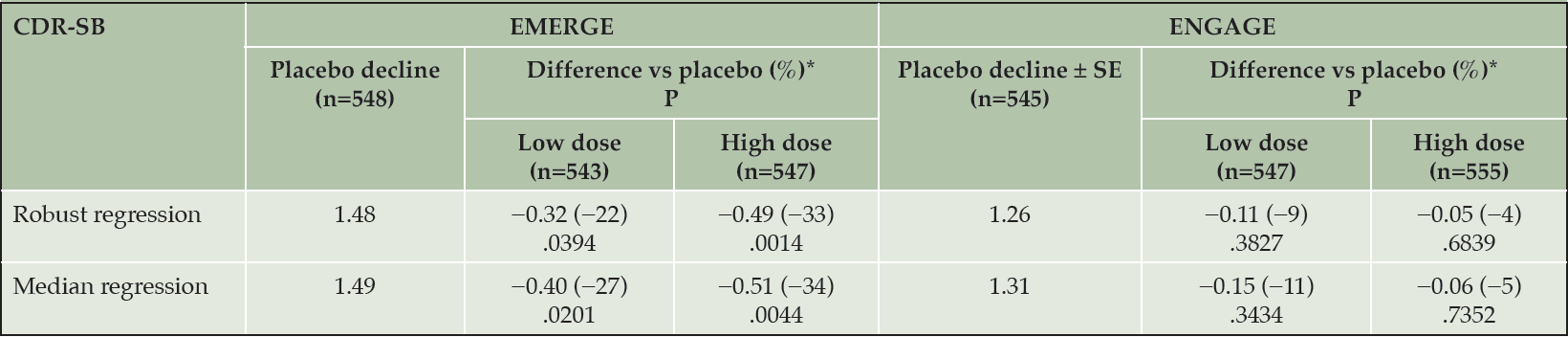
Table 1. Robust and quantile regression analyses
Missing data imputed assuming missing at random. Models included treatment group, baseline CDR-SB, baseline MMSE, symptomatic medication use at baseline, region, and laboratory ApoE ε4 status as covariates. Robust regression used MM estimation. *Difference vs placebo at week 78. Negative percentage indicates less progression in the treated arm. ApoE, apolipoprotein E; CDR-SB, Clinical Dementia Rating Scale–Sum of Boxes; MMSE, Mini-Mental State Examination.
Dosing/exposure to aducanumab
In EMERGE and ENGAGE, patients were stratified by ApoE ε4 carrier status to receive low-dose aducanumab, high-dose aducanumab, or placebo. Prior to protocol amendments, the low-dose group was titrated to a target dose of 3 mg/kg (ApoE ε4+) or 6 mg/kg (ApoE ε4–) while the high-dose group was titrated to a target dose of 6 mg/kg (ApoE ε4+) or 10 mg/kg (ApoE ε4–). Data from the proof-of-concept study (PRIME), which was available in the fall of 2016 (after the start of EMERGE and ENGAGE), suggested that the incidence of ARIA among ApoE ε4 carriers titrated to 10 mg/kg was lower than in patients who received fixed-dose 10 mg/kg (12). Therefore, an amended protocol version 4 (PV4) changed the target dose after titration for ApoE ε4 carriers from 6 to 10 mg/kg. Because EMERGE started later than ENGAGE, EMERGE enrolled 200 more patients after the amendment than ENGAGE; therefore, more patients in EMERGE had the opportunity to receive the full 10 mg/kg target dose (3).
The impact of the PV4 amendment on dosing was assessed in post-hoc analyses by summarizing dosing by enrollment cohorts of every 200 patients. In early-enrolled patients, mean number of doses of 10 mg/kg was lower in ENGAGE vs EMERGE (first 200 patients: 1 vs 4; patients 201–400: 5 vs 7; patients 401–600: 10 vs 11). As the PV4 amendment was implemented over time across the 348 investigative sites in 20 countries, exposure to 10 mg/kg increased substantially, with similar exposures in the 2 studies among later-enrolled patients (Figure 1).
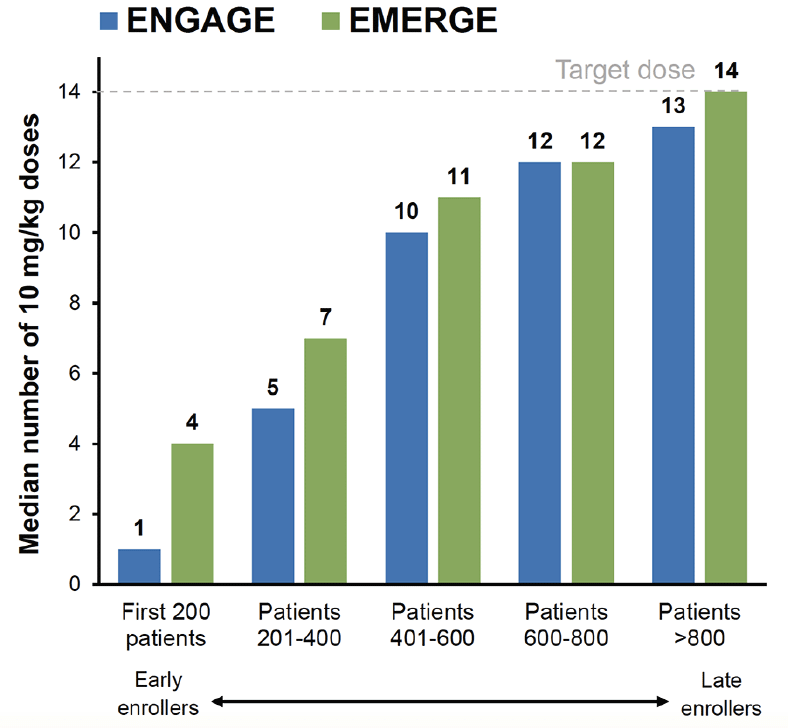
Figure 1. Graph summarizing the median number of
10 mg/kg doses received by each enrollment cohort of 200 over time for EMERGE and ENGAGE
Data included patients who had the opportunity to complete the week 78 visit by March 20, 2019.
The potential impact of the change in dosing that resulted from the PV4 amendment was assessed by excluding early enrolled patients progressively by enrollment cohorts consistent with the dosing cohorts shown in Figure 1. Mean differences from placebo on the CDR-SB, MMSE, ADAS-Cog13, and ADCS-ADL-MCI using this analysis are summarized in Figure 2. Excluding the first 800 enrolled patients excluded most of the patients enrolled under the pre-PV4 dosing regimens. For CDR-SB, results from the high-dose arms of the later-enrolled patients were similar between the 2 studies. Mean differences from placebo showed 29% and 24% slowing of decline in ENGAGE and EMERGE, respectively. The trend for greater mean difference from placebo with increased exposure to 10 mg/kg in later-enrolled patients was stronger and more consistent in ENGAGE. Similar trends were observed for the MMSE, ADAS-Cog, and ADL (Figure 2).
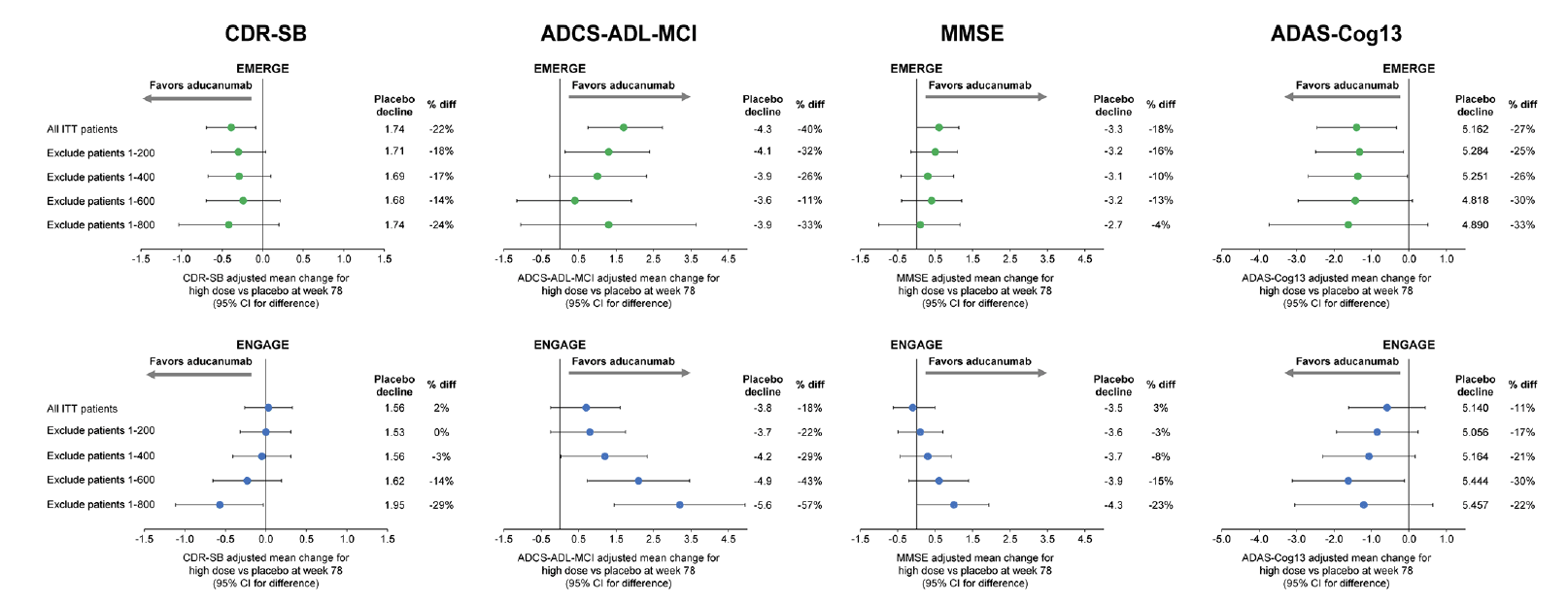
Figure 2. Mean differences between high-dose aducanumab and placebo on primary and secondary endpoints by enrollment cohorts of 200 patients
ADAS-Cog13, Alzheimer’s Disease Assessment Scale–Cognitive Subscale, 13 items; ADCS-ADL-MCI, Alzheimer’s Disease Cooperative Study–Activities of Daily Living Inventory, mild cognitive impairment version; CDR-SB, Clinical Dementia Rating Scale–Sum of Boxes; diff, difference; ITT, intention to treat; MMSE, Mini-Mental State Examination.
Data included patients who had the opportunity to complete the week 78 visit by March 20, 2019.
In addition, the patients who consented to PV4 early enough in study treatment to have full access to 10 mg/kg dosing (PV4 subset) were compared with the patients who did not have this same opportunity for the full 14-dose regimen of 10 mg/kg (pre-PV4 subset). Clinical outcomes in these subsets were compared using the prespecified analyses described in the primary report (3). Mean differences from placebo on CDR-SB for the 4 subsets defined by ApoE ε4 carriage (carrier, noncarrier) and PV4 (pre-PV4, PV4) in the high-dose arms are summarized in Figure S3. Patients with the opportunity to receive all 14 doses of 10 mg/kg (all ApoE ε4 noncarriers and PV4 ApoE ε4 carriers) showed similar mean differences from placebo in the 2 trials. Thus, the difference in results between the high-dose arms was driven by pre-PV4 ApoE ε4 carriers as these patients did not have the opportunity to receive the target dosing regimen.
The impact of dosing, however, was confounded by the impact from the imbalance in rapidly progressing patients in ENGAGE. In the ENGAGE high-dose group, 8 of the 9 rapid progressors with respect to CDR-SB (change >8) over 78 weeks were in the first 800 enrolled patients. Hence, the evolution of mean differences vs placebo in ENGAGE reflected both increased dosing and mitigation of the effects from the imbalance in rapidly progressing patients as the number of extreme observations became more evenly spread across treatment groups. In EMERGE, in which rapid progressors were distributed evenly over time and across treatment arms, later-enrolled patients had a slightly larger difference vs placebo (24%) than in all randomized patients (22%), which may provide a better estimate of the individual contribution of increased dosing to the evolution of differences vs placebo over time.
Synthesizing evidence across studies
After ascertaining the major factors that caused the difference between EMERGE and ENGAGE, results were synthesized across studies to understand the totality of the data. Synthesis was performed in 2 ways. First, a pooled analysis was conducted by combining data from the 2 phase 3 studies and applying the primary analysis model, to which one term was added to account for individual study effects. Then, a plot of mean difference from placebo in CDR-SB and Aβ positron emission tomography (PET) standardized uptake value ratio (SUVR) from all dose arms of the 2 phase 3 studies and the proof-of-concept study (PRIME) (2, 3) was created. This plot (published as Supplemental Data Fig. 4b in Budd Haeberlein S, et al. 2022) illustrates the dose-response relationship for each outcome and the correlation between the outcomes (3).
Figure 3 summarizes mean differences vs placebo for the high-dose arm using pooled data from EMERGE and ENGAGE. The high-dose treatment effects were between those of the individual studies, as expected. The ADAS-Cog13 and ADCS-ADL-MCI were (nominally) significant in the intention-to-treat (ITT) data set, with 19% and 30% reductions, respectively, in clinical decline compared with placebo. The subset of patients who were randomized to receive the target dose of 10 mg/kg (i.e., the PV4 subset) is especially relevant because this is the approved dosing regimen for aducanumab. The PV4 subset approximately corresponds to excluding the first 800 enrolled patients. Larger treatment effects were observed on the CDR-SB, MMSE, and ADCS-ADL-MCI in the pooled PV4 subset than in the all-patient cohorts that included pre-PV4 patients. Results on the ADAS-Cog13 were similar in the all-patient and PV4 cohorts. A 23% slowing of decline was observed on the primary outcome, CDR-SB, with smaller treatment effects on MMSE and ADAS-Cog, and 47% slowing of decline on functional outcomes (ADCS-ADL-MCI).
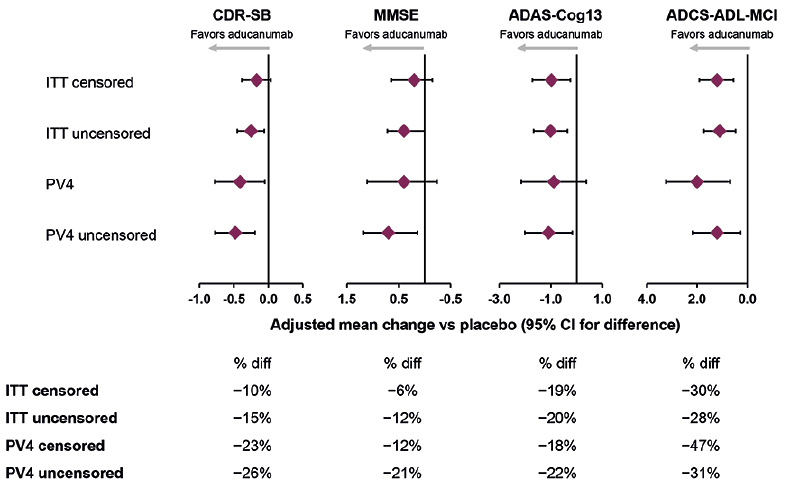
Figure 3. Pooled studies EMERGE and ENGAGE: results in the high-dose group on primary and secondary endpoints at week 78
“ITT censored” describes all data collected under protocol-specified double-blind conditions, with data censored after the announcement of futility. “ITT uncensored” is nearly identical to the ITT censored population, but with approximately 100 additional observations per study coming from the safety follow-up assessment that occurred after futility declaration. Thus, in these follow-up visits, treatment assignment is no longer fully blinded. “PV4 censored” includes all the data collected under protocol-specified double-blind conditions, with data censored after the announcement of futility, on patients randomized early enough in the study to have full access to the targeted treatment regimen. “PV4 uncensored” was the same as the PV4 data set but with data collected after the futility declaration was included, which were no longer fully blinded to treatment assignment. ADAS-Cog13, Alzheimer’s Disease Assessment Scale–Cognitive Subscale, 13 items; ADCS-ADL-MCI, Alzheimer’s Disease Cooperative Study–Activities of Daily Living Inventory, mild cognitive impairment version; ApoE, apolipoprotein E; CDR-SB, Clinical Dementia Rating Scale–Sum of Boxes; diff, difference; ITT, intention to treat; MMSE, Mini-Mental State Examination; PV4, protocol version 4.
Changes from baseline in PET SUVR in the amyloid PET population have been published alongside CDR-SB data for dose arms in the 3 placebo-controlled studies (PRIME, ENGAGE, and EMERGE) (2, 3). In all of these studies, increased dose was associated with both increased mean amyloid removal in the brain and greater mean slowing of clinical decline. The high-dose arm in ENGAGE is the only group that deviated from the overall trend. Given the dosing and amyloid removal, CDR-SB results for high-dose ENGAGE should have been intermediate to the low-dose groups of both studies and the high-dose group of EMERGE. When analysed after accounting for differences in exposure, as measured by cumulative dose or area under the curve, the high-dose arm in EMERGE and ENGAGE demonstrated comparable plaque removal (13).
“ITT censored” describes all data collected under protocol-specified double-blind conditions, with data censored after the announcement of futility. “ITT uncensored” is nearly identical to the ITT censored population, but with approximately 100 additional observations per study coming from the safety follow-up assessment that occurred after futility declaration. Thus, in these follow-up visits, treatment assignment is no longer fully blinded. “PV4 censored” includes all the data collected under protocol-specified double-blind conditions, with data censored after the announcement of futility, on patients randomized early enough in the study to have full access to the targeted treatment regimen. “PV4 uncensored” was the same as the PV4 data set but with data collected after the futility declaration was included, which were no longer fully blinded to treatment assignment. ADAS-Cog13, Alzheimer’s Disease Assessment Scale–Cognitive Subscale, 13 items; ADCS-ADL-MCI, Alzheimer’s Disease Cooperative Study–Activities of Daily Living Inventory, mild cognitive impairment version; ApoE, apolipoprotein E; CDR-SB, Clinical Dementia Rating Scale–Sum of Boxes; diff, difference; ITT, intention to treat; MMSE, Mini-Mental State Examination; PV4, protocol version 4.
Discussion
While many results were consistent between EMERGE and ENGAGE, the high-dose arms results were discordant. High-dose aducanumab demonstrated significant treatment effects across primary and secondary endpoints in EMERGE, but not in ENGAGE. Mean differences from placebo on biomarkers and clinical outcomes were similar for the low-dose arms, and were intermediate to the results for the high-dose arm in EMERGE (3). For a factor to substantially contribute to the divergence in results between the high-dose arms, that factor must have an appreciable influence on outcomes and be substantially unbalanced across treatment arms and studies.
Baseline demographic and disease characteristics were similar between studies and treatment arms and thus did not substantively contribute to the discordant results between the high-dose arms. The frequency, severity, and management of ARIA also did not differ between the studies. Despite the evident dose exposure differences between the high-dose groups in EMERGE and ENGAGE, the incidence of ARIA was not expected to differ due to the observed characteristics of ARIA within the studies. ARIA tended to occur early in the course of treatment (3, 11) and, for patients in the high-dose arms, this would mean ARIA tended to occur before titration to the target dose of 10 mg/kg was reached. No evidence of systematic bias from potential functional unblinding due to ARIA was evident. These findings suggest that ARIA did not contribute to the difference in results between studies.
Factors that contributed to the divergence in results between the high-dose arms included the imbalance across treatment arms in the number of rapidly progressing patients, and lower exposure to 10 mg/kg dosing in ENGAGE. Although it was not possible to cleanly separate the effects of dosing from the imbalance in the number of rapidly progressing patients because the lower dosing and the imbalance in the small number of rapid progressors in ENGAGE both occurred in early-enrolled patients, the post-hoc analyses suggest that the imbalance in rapid progressors played a major role in the discordance of trial outcomes. The average progression in a rapidly progressing patient (a change in score of 8 units or above) is 9.67. The relative impact of a rapid progressing patient on the mean difference between treatment (~8 units) is more than 20-fold greater than the average mean patient (0.39); that is, one rapid progressor offset the average treatment benefit in 20 patients.
In later-enrolled patients (i.e., the PV4 subset), excluding the first 800 enrolled, results from the high-dose arms of the 2 studies was similar, showing 23% and 29% slowing of decline on the CDR-SB in EMERGE and ENGAGE, respectively. Similar trends were seen on secondary outcomes. The PV4 subset, which comprises predominantly later-enrolled patients, is meaningful because these patients were randomized to receive the target dosing regimen and the number of rapidly progressing patients was balanced across treatment arms. Results from the PV4 subset pooled across the 2 studies showed a 23% slowing of decline on the primary CDR-SB outcome, which was similar to the target of 25% slowing upon which sample size and powering of the phase 3 studies was based. In the ITT uncensored data set, which included efficacy assessments taken at the safety follow-up visit, up to 18 weeks after the last dose of study drug, differences from placebo ranged from 21% to 31% across the 4 clinical outcomes, with each being (nominally) significant.
The divergence in high-dose arm results, and the potential explanations for that divergence included in this paper, should be considered in the context of the circumstances that led to these results. Early termination of the trials due to assumptions in the futility analysis that did not hold (3) resulted in fewer patients completing the trials than anticipated. The implementation of protocol amendments based on learnings from the proof-of-concept study (2) increased the exposure for two-thirds of the high-dose arm to 10 mg/kg dosing in the middle of the trial. This in turn increased the overall heterogeneity in the data and, due to the differences between studies in enrollment timing relative to implementation of the amendments, also contributed to the divergence in results of the high-dose arms.
The EMERGE and ENGAGE trials provide a learning opportunity to help inform the design and execution of clinical trials in Alzheimer’s disease. Key points include the need to maximize the “currentness” of the data used in interim analyses; that is, the amount of data collected but not included in the interim analysis should be minimized. Analytic alternatives to MMRM that are less immune to the influence of unusual patients (e.g., rapid progressors) should be evaluated. Trialists should carefully consider the potential impact on data heterogeneity from changes in design, such as dosing, to an ongoing trial. As difficult as it is to delay a trial, it may be better to wait while an important design issue is being resolved (e.g., via data from another trial).
Importantly, limitations of this work should be considered. The present investigation relied on post-hoc analyses, and thus results should be interpreted in that light. To mitigate these limitations, the hypothesis and analytical approaches were specified before analyses were conducted. Additionally, it is important to view these results as a partial explanation for the discordance in results between the high-dose arms, and not as a replacement for the prespecified, primary study results in which ENGAGE did not show a treatment effect.
Overall, findings were consistent across studies in later-enrolled patients, among which the incidence of rapidly progressing patients was balanced across treatment arms.
Funding: The sponsor (Biogen) played a role in the design and conduct of these studies as well as the collection, analysis, and interpretation of data. Medical writing and editorial support were provided by MediTech Media, Ltd., in accordance with Good Publication Practice guidelines (http://www.ismpp.org/gpp-2022) and were funded by Biogen.
Acknowledgments: The authors thank the participants and their family members in the aducanumab studies as well as the investigators and their staff conducting these studies.
Author Contributions: All authors made substantial contributions to the acquisition, analysis, or interpretation of the data; critically revised the manuscript for intellectual content; approved the final version to be published; and are accountable for all aspects of the work.
Competing Interest Declaration: These studies were funded by Biogen. YT, GD, KH, KKM, IN, SW, YZ, TC, and SBH are employees and shareholders of Biogen. CvH, PB, CM, LN, RR, and AS are former employees of Biogen. CM is a current employee of Cortexyme. PSA was Chair of the Steering Committee; has received research support from Eisai, Eli Lilly, Janssen, the Alzheimer’s Association, NIH, and FNIH; and has consulted for ImmunoBrain Checkpoint, Merck, and Roche. FB was supported by the NIHR Biomedical Research Centre at UCLH. He receives personal fees for consultancy from Bayer, Biogen, Combinostics, IXICO, Novartis, and Roche. SC was an ENGAGE trial site investigator and an Aducanumab Steering Committee member. She is a consultant to Alnylam, Biogen, Cognivue, Cogstate, Eisai, Eli Lilly, INmune Bio, Novo Nordisk, ProMIS Neurosciences, RetiSpec, and Roche and receives research support (paid to institution) from AgeneBio, Alector, Alnylam, Anavex, Biogen, Eisai, Eli Lilly, Genentech, Novo Nordisk, RetiSpec, Roche, UCB, and Vielight. OH has acquired research support (for the institution) from Avid Radiopharmaceuticals, Biogen, Eisai, Eli Lilly, GE Healthcare, Pfizer, and Roche. In the past 2 years, he has received consultancy/speaker fees from AC Immune, ALZPath, Biogen, Cerveau, and Roche. TI is a consultant to Eisai and Roche. CJM was an ENGAGE trial site investigator and an Aducanumab Steering Committee member. She is supported by the NIHR Biomedical Research Centre at UCLH and has acted as a consultant to Biogen, Ionis, and Roche. CvD was an EMERGE trial site investigator. He is a consultant to Eisai and Roche and receives research support from Biogen, Biohaven, Eisai, Eli Lilly, Genentech, Janssen, Merck, Novartis, and Roche. BV was a trial site investigator and an Aducanumab Steering Committee member. He is supported by CHU-T and has acted as a consultant to Biogen, Eli Lilly, Merck, and Roche.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. Arndt, J.W. et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci Rep. 8, 6412 (2018). https://doi.org/10.1038/s41598-018-24501-0
2. Sevigny, J. et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 537, 50–56 (2016). https://doi.org/10.1038/nature19323
3. Budd Haeberlein, S. et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J Prev Alzheimers Dis 9, 197–210 (2022). https://doi.org/10.14283/jpad.2022.30
4. Linse, S. et al. Kinetic fingerprints differentiate the mechanisms of action of anti–Aβ antibodies. Nat Struct Mol Biol. 27, 1125–1133 (2020). https://doi.org/10.1038/s41594-020-0505-6
5. Aduhelm. Prescribing information. Biogen, Inc.; 2021.
6. Holland, P. W., and Welsch, R. E. Robust regression using iteratively reweighted least-squares. Communications in Statistics—Theory and Methods (1977). https://doi.org/10.1080/03610927708827533
7. Huber, P. J. Robust Regression: Asymptotics, conjectures, and Monte Carlo. Ann Statist (1973). 1:799–821. https://doi.org/10.1214/aos/1176342503
8. SAS Institute Inc. SAS/STAT® 15.1 User’s Guide. Cary, NC: SAS Institute Inc (2018).
9. Koenker, R., and Bassett, G. W. Regression quantiles. Econometrica (1978). https://doi.org/10.2307/1913643
10. Sperling, R.A. et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 7, 367–385 (2011). https://doi.org/10.1016/j.jalz.2011.05.2351
11. Salloway S, et al. Amyloid-related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol. 79, 13–21 (2021). doi:10.1001/jamaneurol.2021.4161
12. Budd Haeberlein, S. et al. Aducanumab titration dosing regimen: 36-month analyses from PRIME, a Phase 1b study in patients with early Alzheimer’s disease. Platform presentation. CTAD 2018.
13. Muralidharan K, et al. Population pharmacokinetics and standard uptake value ratio of aducanumab, an amyloid plaque-removing agent, in patients with Alzheimer’s disease. CPT Pharmacometrics Syst Pharmacol. Online ahead of print. (2021). https://doi: 10.1002/psp4.12728.
© The Authors 2023