
E. Teng1, P.T. Manser1, M. Shah1, K. Pickthorn1, N. Hu1, S. Djakovic1, H. Swendsen1, M. Blendstrup1, G. Faccin2, S. Ostrowitzki2, K.M. Sink1
1. Genentech, Inc., South San Francisco, CA, USA; 2. F. Hoffmann-La-Roche Ltd., Basel, Switzerland.
Corresponding Author: Edmond Teng, MD, PhD, Genentech, Inc., 1 DNA Way, South San Francisco, CA 94080, Tel: 650-467-1661, Fax: 650-467-2887, teng.edmond@gene.com
J Prev Alz Dis 2023;1(10):41-49
Published online November 22, 2022, http://dx.doi.org/10.14283/jpad.2022.101
Abstract
Background: Screening procedures for early Alzheimer’s disease (AD) trials seek to efficiently identify participants who fulfill clinical and biomarker criteria for AD and enrich for those most likely to experience significant clinical progression during the study. Episodic memory performance is often assessed in screening, but the utility of different memory tests for optimizing screening efficiency and/or rates of clinical progression remains uncertain.
Objectives: Cross-study comparisons of the effects of inclusion criteria based on performance on the Free and Cued Selective Reminding Test (FCSRT) or the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) on screen-failure rates for episodic memory and β-amyloid (Aβ) positivity (by CSF or PET) and on subsequent rates of clinical disease progression in randomized participants across three clinical trials in early (prodromal-to-mild) AD.
Design: Secondary analyses of cross-sectional and longitudinal clinical trial data.
Setting: Multi-center international clinical trials.
Participants: Individuals with prodromal-to-mild AD screened and/or randomized in clinical trials for crenezumab (CREAD, CREAD2) or semorinemab (Tauriel). Cross-sectional analyses of screening data for episodic memory impairment included participants from CREAD2 (n=2897) and Tauriel (n=887) and for Aβ positivity included participants from CREAD (n=1138), CREAD2 (n=1119), and Tauriel (n=483). Longitudinal analyses of rates of clinical progression included participants from CREAD (n=779), CREAD2 (n=773), and Tauriel (n=331).
Measurements: Cross-sectional analyses examined eligibility rates per cutoffs defined for the FCSRT (CREAD, CREAD2) or RBANS (Tauriel) and per Aβ positivity using CSF and/or PET biomarkers. Longitudinal analyses examined rates of clinical progression on the Clinical Dementia Rating-Sum of Boxes (CDR-SB), the 13-item version of the Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-Cog13), and Alzheimer’s Disease Cooperative Study-Activities of Daily Living Scale (ADCS-ADL).
Results: Lower rates of study eligibility per episodic memory criteria were seen with the FCSRT (CREAD2) relative to the RBANS (Tauriel), but similar rates of eligibility per Aβ positivity criteria were seen amongst participants with episodic memory impairment per the cutoffs used on either assessment. Similar rates of clinical decline over 18 months on the CDR-SB, ADAS-Cog13, and ADCS-ADL were observed in study populations enriched using the FCSRT (CREAD, CREAD2) or the RBANS (Tauriel).
Conclusions: Cutoffs for episodic memory impairment on the FCSRT used in the CREAD and CREAD2 studies are more stringent than those on the RBANS used in the Tauriel study, resulting in lower rates of eligibility. However, given that study enrichment with either test yields similar rates of Aβ positivity and clinical progression, considerations beyond these factors may drive the decision of which assessment to use for screening in early AD clinical trials.
Key words: Alzheimer, memory, amyloid, clinical trial, screening, inclusion/exclusion.
Introduction
Over the past decade, clinical trials in Alzheimer’s disease (AD) have increasingly focused on enrolling participants at earlier stages of disease, when putative disease-modifying interventions may be more likely to deliver greater clinical impact (1). Clinical trials in early AD entail a number of distinct challenges (2) and rely on efficient screening procedures to identify participants that: a) fulfill clinical diagnostic criteria for AD, b) have biomarker evidence of underlying AD pathology, and c) will exhibit sufficient clinical progression to allow for detection of any treatment effects over the duration of the study.
In the majority of recent early (prodromal-to-mild) AD clinical trials, these screening imperatives have resulted in inclusion criteria that specify significant impairment in episodic memory. Deficits in episodic memory are a core feature of the IWG-2 clinical diagnostic criteria for AD (3). In cross-sectional studies of amnestic mild cognitive impairment (MCI), more severe episodic memory deficits are seen amongst participants who are β-amyloid (Aβ) PET positive than those who are Aβ PET negative (4, 5). Furthermore, longitudinal studies indicate that participants with greater impairment on episodic memory tests at baseline show more rapid subsequent clinical decline (6, 7).
Two episodic memory tests commonly implemented for screening in early AD trials are the Free and Cued Selective Reminding Test (FCSRT) (8) and the Delayed Memory Index (DMI) from the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) (9). The FCSRT is a word-list learning task that specifically assesses the effects of cueing for assisting verbal recall; deficits in cued recall in the context of controlled encoding have been postulated to be more specific for AD (3, 10, 11). The RBANS includes 12 cognitive subtests that assess a wider range of cognitive function. The RBANS DMI is comprised of 4 subtests that explicitly assess delayed memory (Word List Recall, Word List Recognition, Story Memory, and Figure Recall); average performance across these subtests has been postulated to be a more reliable measure of anterograde memory than any single test (12). The FCSRT has been used in trials of crenezumab (NCT02670083, NCT03114657), gantenerumab (NCT01224106), and solanezumab (NCT02760602), while the RBANS DMI has been used in trials of aducanumab (NCT02477800, NCT02484547), semorinemab (NCT03289143), tilavonemab (NCT02880956), and verubecestat (NCT01953601).
Previous work suggests that cutoffs on the FCSRT produce study cohorts that exhibit faster rates of clinical progression than similar unenriched cohorts (13). Likewise, poorer scores on the FCSRT (14) and the RBANS DMI (15) have been associated with higher rates of positive Aβ PET scans. However, the relative utility of the FCSRT versus the RBANS for optimizing screening efficiency and/or rates of subsequent clinical progression in randomized participants remains uncertain. We sought to address these knowledge gaps by comparing study eligibility rates per inclusion criteria for episodic memory deficits and positive Aβ biomarker status as well as subsequent rates of clinical decline between early AD trials that used cutoffs on either the FCSRT (CREAD, CREAD2; crenezumab) or the RBANS DMI (Tauriel; semorinemab).
Methods
Study design
Screening and/or longitudinal data from participants with prodromal-to-mild AD in the CREAD (NCT02670083), CREAD2 (NCT03114657), and Tauriel (NCT03289143) trials were analyzed. The Phase 3 CREAD (n=813) and CREAD2 (n=806) studies evaluated the safety and efficacy of monthly doses of the anti-Aβ antibody crenezumab (60 mg/kg) versus placebo for up to 105 weeks (16), while the Phase 2 Tauriel study (n=457) evaluated the safety and efficacy of monthly doses of the anti-tau antibody semorinemab (1500 mg, 4500 mg, or 8100 mg) versus placebo over 73 weeks (17). As previously reported, no clinical efficacy was seen in these studies with either intervention relative to placebo (16, 17).
Participants
At the time of enrollment, participants in all three studies met diagnostic criteria for mild cognitive impairment (MCI; i.e., prodromal AD) (18) or dementia (mild AD dementia; i.e., mild AD) (19) due to AD, and had global Clinical Dementia Rating (CDR) (20) scores of 0.5 or 1, and evidence of significant cerebral amyloid pathology confirmed by Aβ PET scan (visual read of [18F]florbetaben, [18F]florbetapir, or [18F]flutemetamol in all studies, also [18F]NAV4694 in Tauriel (21-24)) or CSF Aβ1-42 levels [CREAD, CREAD2: ≤ 950 pg/mL Tauriel: ≤ 1000 pg/mL, Elecsys β-amyloid (1-42) CSF immunoassay; Roche Diagnostics, Penzberg, Germany; cutoffs differ in part due to different CSF sample handling protocols]. The inclusion criteria for the crenezumab and semorinemab studies differed in age (CREAD, CREAD2: 50-85 years old; Tauriel: 50-80 years old) and Mini-Mental Status Examination (MMSE) (25) score [CREAD, CREAD2: 22-30 (inclusive); Tauriel: 20-30 (inclusive)] ranges. In order to increase the validity of comparisons between studies, only participants with screening MMSE scores of 22-30 (inclusive) were included in our analyses.
For the episodic memory deficit inclusion criterion, the CREAD and CREAD2 studies used cutoffs derived from the FCSRT (8) Immediate Recall assessments (Free Recall ≤ 27 and Cueing Index ≤ 0.67) which had previously been identified as optimal for enriching prodromal AD studies for participants most likely to demonstrate subsequent clinical progression (26). The FCSRT is estimated to take 12-15 minutes to administer (27). In the Tauriel study, the analogous inclusion criterion used a cutoff derived from the RBANS (9) [DMI ≤ 85 (one standard deviation below normative means)]. The RBANS is estimated to take 20-30 minutes to administer (28). Psychometric analyses (including test-retest reliability) have previously been reported for both the FCSRT (29) and the RBANS (28, 30). Centralized service providers (CREAD and CREAD2: Bracket, Wayne, PA; Tauriel: Medavante-ProPhase, Hamilton Township, NJ) were used to standardize rater qualifications and training, test administration, data quality monitoring, and scoring of the FCSRT and RBANS across study sites via tablet-based electronic data capture.
Screening procedures
The screening procedures for CREAD, CREAD2, and Tauriel were performed in stages, as illustrated in Supplemental Figure 1. MMSE and episodic memory (FCSRT or RBANS) assessments were performed during the initial screening stage. Comparisons of eligibility per episodic memory impairment inclusion criteria were only conducted with CREAD2 and Tauriel data because in CREAD, the FCSRT was often performed prior to the MMSE. Amongst participants screened for CREAD who did not meet the FCSRT inclusion criterion, MMSE scores were not consistently collected. Across all three studies, if MMSE and episodic memory criteria were met, participants proceeded to the next stage of screening, which included assessment with the CDR, diagnostic classification as prodromal or mild AD, medical history and examination, laboratory assessments, and brain MRI. Determination of amyloid biomarker status positivity by CSF and/or PET was performed at the last stage of screening for potential participants who fulfilled all other inclusion and exclusion criteria. A subset of participants underwent both CSF and PET assessments for amyloid status (CREAD: n=112; CREAD2: n=22; Tauriel: n=41) either because they elected to undergo additional optional study procedures or because their amyloid status was negative in the first modality assessed (either PET or CSF) and they elected to be re-assessed with the other modality. The overall screen-failure rates for each study have previously been reported (CREAD: 78.2%; CREAD2: 78.0%, Tauriel 63.1%) (16, 17).
Longitudinal clinical outcome assessments
The schedule of overlapping longitudinal clinical outcome assessments for CREAD, CREAD2, and Tauriel are illustrated in Supplemental Figure 2. All studies included assessments of the CDR Sum of Boxes (CDR-SB), the ADAS-Cog13 (31), and Alzheimer’s Disease Cooperative Study-Activities of Daily Living Scale (ADCS-ADL) (32). Assessments were performed at Weeks 1 and 25 in all three studies, with additional assessments performed at Weeks 53, 77, and 105 in CREAD and CREAD2, and at Weeks 49 and 73 in Tauriel. For consistent comparisons between studies, the analyses reported here only included assessments up to Weeks 73/77. The Tauriel study was conducted through the completion of its double-blind period (Week 73; mean duration of treatment: 62.0 weeks), but the CREAD and CREAD2 studies were halted early after an interim futility analysis (16). More longitudinal data was available from CREAD (mean duration of treatment: 78.8 weeks) than from CREAD2 (mean duration of treatment: 41.9 weeks). For each clinical outcome measure, participants included in the respective modified intent-to-treat (mITT) cohort had assessment data at baseline and at least one post-baseline timepoint.
Statistical Analyses
Primary statistical analyses were performed with R (v.3.3.2) (33). Baseline comparisons between trial cohorts were conducted using t-tests or one-way ANOVAs for continuous measures and chi-squared tests for categorical variables, with post hoc Bonferroni correction for multiple comparisons where appropriate. Inclusion rates during screening per the episodic memory criterion using the FCSRT (CREAD2) versus RBANS (Tauriel) and per the Aβ positivity criterion using Aβ PET or CSF Aβ1-42 were compared between trial cohorts using chi-squared tests, with Bonferroni correction for multiple comparisons across MMSE bins. Longitudinal rates of disease progression, as measured by the CDR-SB, ADAS-Cog13, and ADCS-ADL were estimated using mixed models for repeated measures (MMRM), adjusting for baseline diagnosis (prodromal versus mild AD), APOE genotype (ε4+ versus ε4-), and baseline performance on each assessment.
Data Sharing
Qualified researchers may request access to individual patient level data through the clinical study data request platform (https://vivli.org/). Further details on Roche’s criteria for eligible studies are available here: https://vivli.org/members/ourmembers/. For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see https://www.roche.com/innovation/process/clinical-trials/data-sharing/.
Results
Screening populations in CREAD2 and Tauriel
Demographic and clinical information for participants screened in CREAD2 and Tauriel who had baseline MMSE scores of 22-30 (inclusive) and available FCSRT (CREAD2) or RBANS (Tauriel) data are shown in Table 1. Due to the different inclusion criteria for age between the two studies, screening participants in CREAD2 were significantly older than those in Tauriel [t(3782)=8.37, p<0.001]. Similar screening MMSE scores were observed in the analysis cohorts for these two studies [t(3782)=1.12, p=0.27].
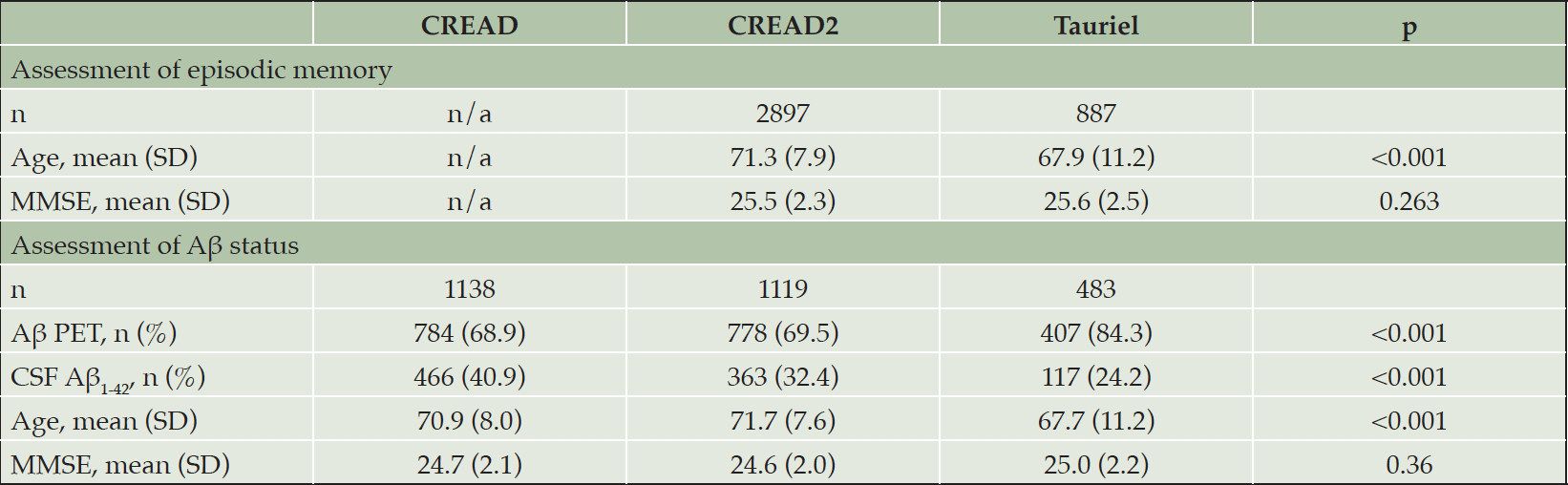
Table 1. Screening cohorts for CREAD2 and Tauriel
The total number of Aβ PET and CSF Aβ1-42 assessments exceeds the total number of participants because a subset of participants was assessed for Aβ using both modalities.
Eligibility per episodic memory impairment inclusion criterion
Eligibility rates for screening participants in CREAD2 and Tauriel per their respective inclusion criteria cutoffs on the FCSRT (CREAD2) or RBANS (Tauriel) are shown in Figure 1. In the overall analysis cohort, significantly higher percentages of screening participants were eligible with the RBANS (76.3%) versus FCSRT (54.6%) cutoffs [χ2(3784)=133.51, p<0.001]. In both CREAD2 and Tauriel, higher eligibility rates per the episodic memory impairment criteria were seen at lower MMSE scores, but for each MMSE score (from 22-30), higher rates of eligibility were seen with the RBANS cutoff relative to the FCSRT cutoff (all ps≤0.001, Bonferroni-corrected critical p=0.006).
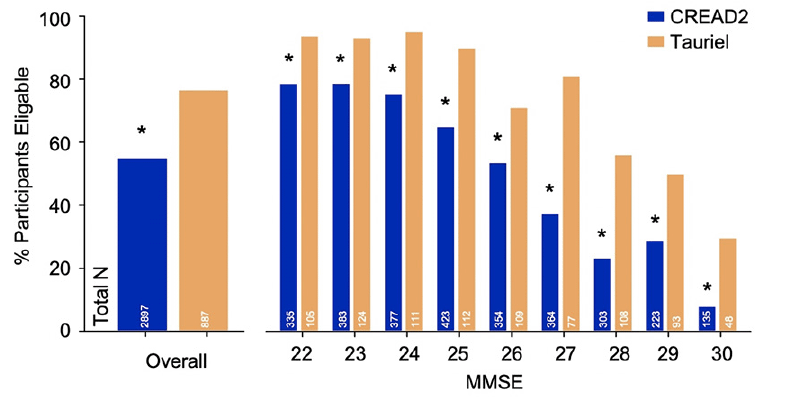
Figure 1. Eligibility rates per episodic memory impairment criteria were lower with FCSRT versus RBANS cutoffs
*p<0.001 vs. RBANS.
Eligibility per Aβ positivity inclusion criterion
In CREAD, CREAD2, and Tauriel, only participants who fulfilled the episodic memory impairment inclusion criterion (and other clinical inclusion/exclusion criteria) were subsequently assessed for Aβ positivity by either PET or CSF. Similar proportions of participants failed screening due to other clinical inclusion/exclusion criteria between MMSE/episodic memory testing and Aβ assessment in CREAD (29.9%), CREAD2 (29.2%), and Tauriel (28.7%). Demographic and clinical information for participants who reached the Aβ assessment stage of screening are shown in Table 1. There was again a significant difference between studies in age [F(2,2737)=38.10, p<0.001], as CREAD and CREAD2 participants who underwent assessment for Aβ positivity were significantly older than those in Tauriel (ps<0.001). Similar MMSE scores were seen across study cohorts [F(2,2737)=1.02, p=0.36]. There were significant differences between trials in the proportions of screening participants with Aβ PET data [χ2(2,2740)=44.72, p<0.001; CREAD: 68.9%, CREAD2: 69.5%, Tauriel: 84.3%]. Likewise, there were significant differences between trials in the proportions of screening participants with CSF Aβ1-42 data [χ2(2,2740)=45.60, p<0.001; CREAD: 40.9%, CREAD2: 32.4%, Tauriel 24.2%].
Similar rates of Aβ PET positivity (Figure 2A) were seen across the three studies [CREAD: 70.4%, CREAD2: 70.8%, Tauriel: 68.3%; χ2(2,1969)=0.85, p=0.65] and across screening MMSE scores (all ps≥0.10), though higher rates were seen in both studies in participants with lower MMSE scores. Likewise, similar rates of CSF Aβ1-42 positivity (Figure 2B) were seen across the three studies [CREAD: 79.6%, CREAD2: 81.8%, Tauriel: 85.5%; χ2(2,946)=2.25, p=0.32] and across screening MMSE scores (all ps≥0.09). However, no clear association relationship between screening MMSE score and CSF Aβ1-42 positivity was observed, possibly due to the smaller sample sizes of participants assessed for Aβ positivity via CSF. Within each study, higher rates of Aβ biomarker confirmation were seen amongst participants assessed via CSF relative to those assessed via PET [CREAD: χ2(1250)=12.82, p<0.001; CREAD2: χ2(1141)=15.68, p<0.001; Tauriel: χ2(524)=13.32, p<0.001]. However, analyses of data from a CREAD substudy that included participants who underwent both CSF Aβ1-42 and Aβ PET analyses demonstrated 85% concordance for Aβ positivity between these modalities (16).
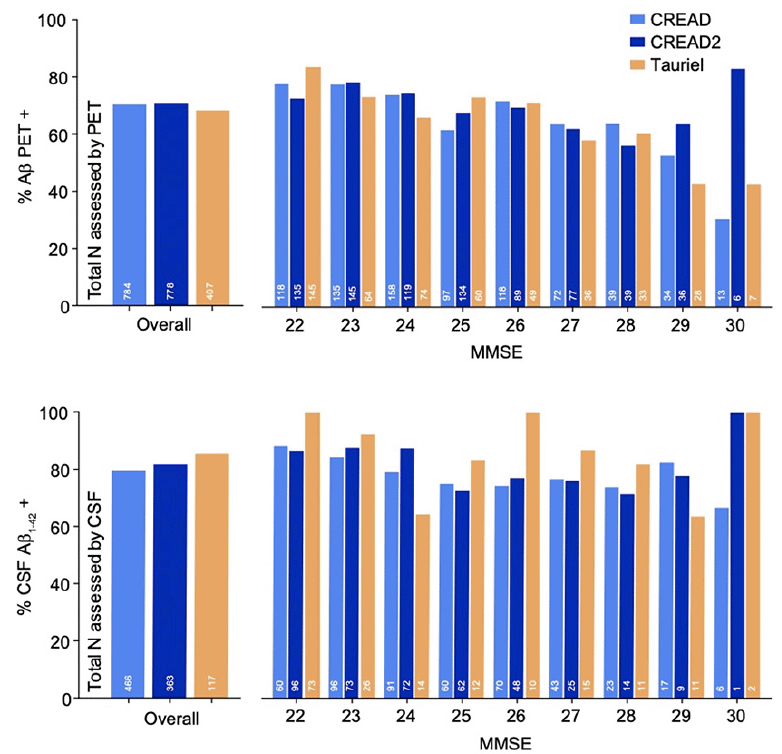
Figure 2. Aβ biomarker positivity rates were not significantly different after enrichment via FCSRT (CREAD2) or RBANS (Tauriel) with A) Aβ PET and B) CSF Aβ1-42 levels
Longitudinal progression rates on clinical outcome assessments
Although previous work suggests that the FCSRT may have utility for enriching early AD clinical trials for participants with positive Aβ PET scans (14), the inclusion cutoff on the FCSRT used in CREAD and CREAD2 was optimized to enrich for early AD participants who were most likely to show subsequent clinical progression rather than for Aβ positivity (26). Therefore, we performed additional analyses of longitudinal disease progression in the CREAD, CREAD2, and Tauriel studies to determine whether the different inclusion criteria for episodic memory impairment between CREAD/CREAD2 and Tauriel resulted in different patterns of clinical decline.
We initially compared disease progression between the placebo arms of the CREAD, CREAD2, and Tauriel studies. Baseline characteristics of these three placebo cohorts are shown in Table 2. While the placebo arms of these studies were generally well balanced, there was a slightly higher percentage of White participants in the Tauriel placebo arm and slightly poorer baseline performance on the ADAS-Cog13 in the CREAD and CREAD2 placebo arms. Longitudinal analyses of change from baseline through Week 73/77 on the CDR-SB, ADAS-Cog13, and ADCS-ADL in the two placebo groups is shown in Figure 3A. MMRM modeling indicated that similar overall rates of clinical decline and variance were observed in across all three studies.
We have previously reported that clinical efficacy was not observed in any of the treatment arms in CREAD, CREAD2, or Tauriel relative to placebo (16, 17). Therefore, in order to compare overall rates of clinical decline between studies, we pooled all of the treatment arms within each study for further analyses. Baseline characteristics of the three overall cohorts are shown in Table 2. While these larger cohorts were again generally well balanced, there was again a higher percentage of White participants in Tauriel. CREAD2 was comprised of a slightly lower percentage of APOE4 carriers, while CREAD was comprised of a slightly higher percentage of mild AD participants. Overall, slightly better performance was seen at baseline on the MMSE, ADAS-Cog13, and ADCS-ADL in Tauriel. MMRM modeling again indicated that both the overall rates of clinical decline and variance were similar across all three studies (Figure 3B).
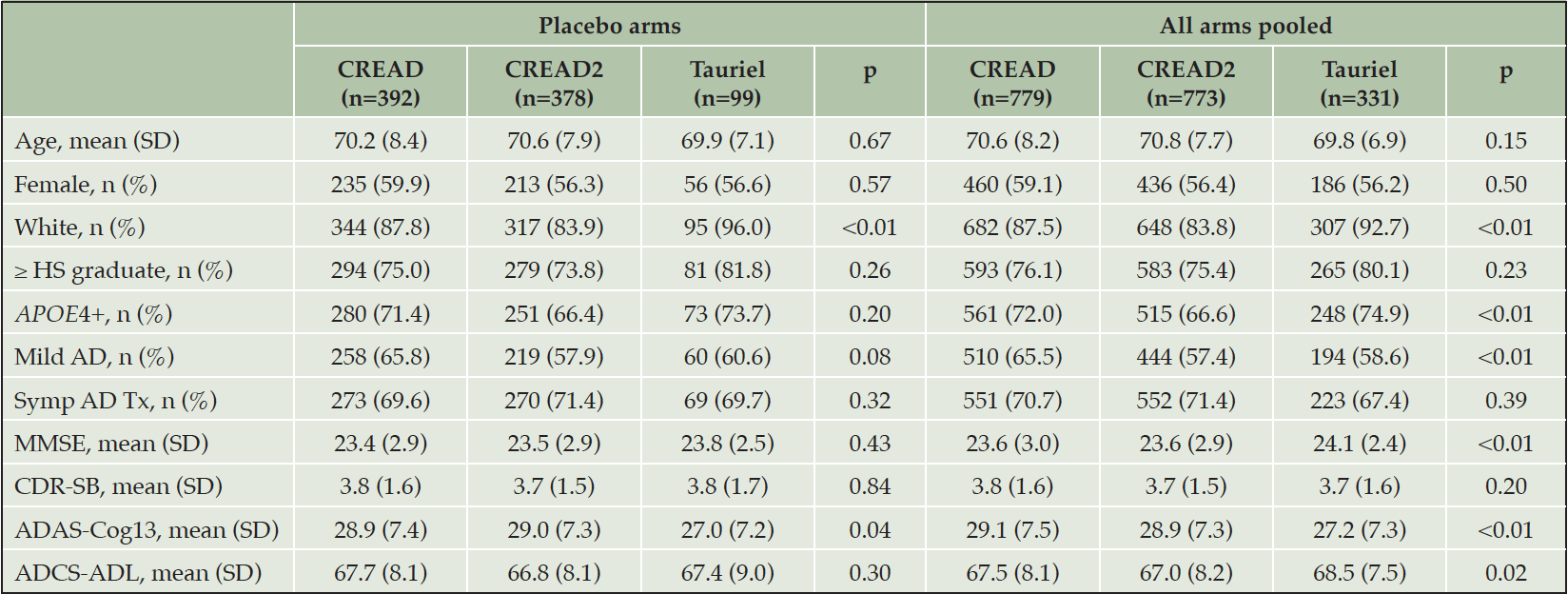
Table 2. Baseline demographics and disease characteristics for modified Intent-to-Treat (mITT) cohorts of the CREAD and Tauriel studies
mITT: participants with CDR-SB (primary endpoint) data at baseline and at least one post-baseline timepoint; HS: high school; Symp AD Tx: symptomatic treatment with an acetylcholinesterase inhibitor and/or memantine.
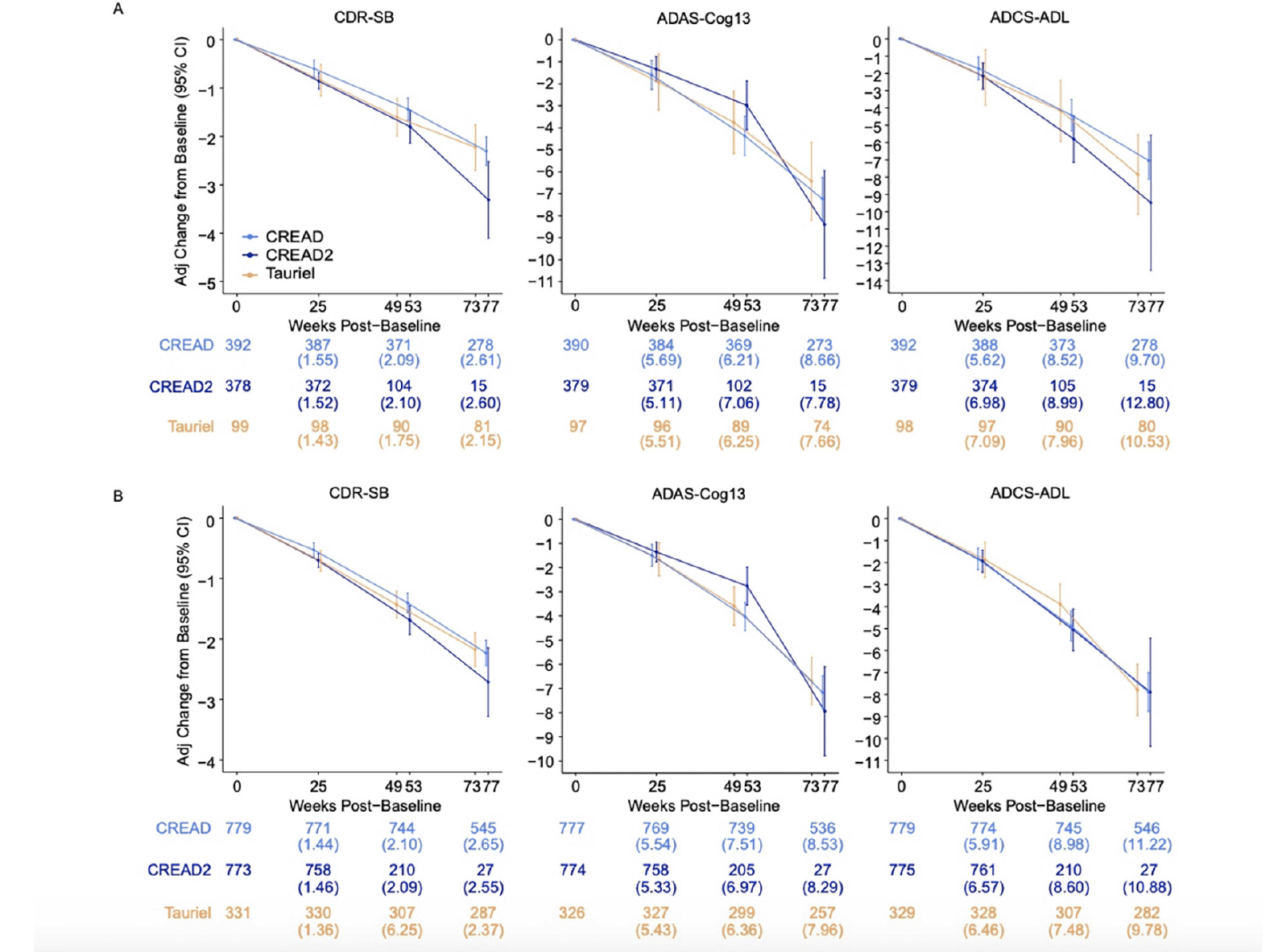
Figure 3. Mixed models for repeated measures (MMRM) least squares estimates of mean change from baseline on the CDR-SB, ADAS-Cog13, and ADCS-ADL were similar after enrichment via FCSRT (CREAD and CREAD2) or RBANS (Tauriel) in A) placebo arms only and B) all arms pooled
Numbers below plots at each timepoint represent the number of patients and (SD).
Discussion
Our analyses of the CREAD, CREAD2, and Tauriel studies suggest that while FCSRT cut-offs resulted in a lower rate of eligibility relative to RBANS cut-offs per episodic memory inclusion criteria, the cohorts that resulted from the use of either test within the overall screening process yielded similar rates of AD biomarker positivity. Likewise, amongst randomized participants, similar rates of subsequent clinical decline were seen on the CDR-SB, ADAS-Cog13, and ADCS-ADL scales. Taken together, these results suggest that the FCSRT and RBANS cutoffs used in these studies exhibit similar utility for enriching the study samples for both positive AD biomarkers and significant longitudinal clinical disease progression.
The lower rates of eligibility per episodic memory inclusion criteria in CREAD2 relative to Tauriel likely reflect that the FCSRT cutoff that was used selects for more severe memory impairment than the corresponding RBANS cutoff, as suggested by the poorer baseline performance on the MMSE and ADAS-Cog13 amongst participants in CREAD and CREAD2 relative to those in Tauriel (Table 2). While there are limited formal FCSRT normative data available for the version that used in CREAD and CREAD2, data from a cognitively normal Italian cohort indicates that a Cueing Index cutoff of ≤0.67 is approximately 2.4 SD below the normative mean (34). In contrast, the RBANS DMI cutoff of ≤85 that was used in Tauriel is only 1 SD below the normative mean. The corresponding cutoff for 2.4 SD below the normative mean would be ≤64. If implemented for Tauriel, this cutoff would decrease the episodic memory criterion eligibility rate to 55.1%, which is comparable to that seen in CREAD2 (54.6%).
A prior analysis of the utility of using an RBANS DMI cutoff of ≤85 for screening across multiple early AD studies reported an Aβ PET positivity rate of 70% (35), which is similar to the Aβ PET positivity rates seen in our cohorts when using either the FSCRT or RBANS inclusion criteria. In comparison, in the Alzheimer’s Disease Neuroimaging Initiative dataset, where cognitively impaired participants (MCI or dementia) were required to demonstrate impairment on the Logical Memory II subscale of the Wechsler Memory Scale-Revised (36), only 57% of Aβ PET scans are positive (37). The relatively modest improvements in rates of positive Aβ PET scans with the FCSRT or RBANS may reflect previous work suggesting that the correlation between Aβ load and episodic memory is attenuated with increasing disease severity (38), given that the majority of participants across the three studies analyzed here were diagnosed with mild AD dementia.
The FCSRT Cueing Index cutoff of ≤0.67 was not intended to optimize enrichment for Aβ positivity but rather to optimize enrollment of prodromal AD patients who are more likely demonstrate subsequent clinical progression (26), and is analogous to a cutoff (≤0.71) that was previously reported to be prognostic for progression from MCI to dementia (39). Preliminary analyses of CREAD data suggest that participants with prodromal AD in the placebo arm of that study exhibited faster rates of progression on the CDR-SB than prior prodromal AD cohorts with less stringent episodic memory inclusion criteria (13). However, comparable rates of disease progression were seen across the CREAD, CREAD2, and Tauriel studies, despite the putatively less stringent cutoff used for the RBANS DMI relative to the FCSRT. These results could suggest that the relationship between the magnitude of episodic memory deficits at baseline and subsequent rates of cognitive decline is non-linear in nature. Beyond a certain threshold, the additional prognostic value of more profound episodic memory impairment may diminish. This hypothesis is supported by analyses limited to participants in Tauriel with baseline RBANS DMI scores ≤ 64 (i.e., 2.4 SD below normative means and comparable to FCSRT threshold in CREAD and CREAD2), which indicate that rates of decline on clinical measures were largely unchanged relative to the larger Tauriel cohort and consistent with those seen in CREAD and CREAD2 (Supplemental Figure 3). Alternatively, many participants with more advanced disease (e.g., mild AD, which comprised the majority of our study cohorts) may already have quite severe episodic memory impairment deficits, given that 79% of the Tauriel participants included in our analyses had screening RBANS DMI scores ≤ 64.
For the three studies included in the current analyses, higher rates of Aβ positivity were seen amongst participants assessed with CSF relative to those assessed with PET, irrespective of which episodic memory test was used in screening. The underlying explanation for this result remains uncertain, particularly since only a small proportion of participants were assessed with both CSF and PET [which showed 85% concordance in CREAD (16)]. The different rates of Aβ positivity seen between these modalities could reflect their differential availability and/or acceptability across the global networks of study sites used in these trials. Alternatively, in a limited sample of MCI participants from AIBL, CSF Aβ1-42 levels yielded a higher rate of Aβ positivity relative to Aβ PET (40), which may reflect prior findings that CSF Aβ1-42 levels turn positive earlier than Aβ PET (41).
While similar study protocols facilitated the comparison of disease progression rates between the CREAD and Tauriel, further comparisons of disease progression rates with other recent clinical trials in early AD with the same screening MMSE range (22-30) are more challenging, due to subtle differences in inclusion/exclusion criteria (including episodic memory impairment criteria) that may impact disease severity at baseline and differences in statistical modeling that may impact estimated rates of disease progression. While point estimates for change in placebo arms on CDR-SB from baseline to Week 73 (Tauriel) or Week 77 (CREAD) were numerically greater than those reported at analogous timepoints from the placebo arms of other early AD trials that incorporated the RBANS DMI (tilavonemab; NCT02880956) (42), Wechsler Memory Scale-IV Logical Memory II (lecanemab; NCT01767311) (43), or the CDR memory box score (gosuranemab; NCT03352557) (44) into their screening processes, baseline MMSE scores in the CREAD and Tauriel placebo arms were also numerically lower than those reported from the other studies.
There are a few factors that may limit the interpretation of these results. The comparisons of eligibility rates and subsequent clinical progression rates for study participants identified using the FCSRT versus RBANS were side-by-side as opposed to head-to-head. Therefore, differences in eligibility rates could have been affected by differences in screening practices between investigators, sites, and/or countries. However, both the partial overlap in each of these factors across studies and the large number of investigators/sites that contributed participants to each study makes this less likely. The RBANS DMI cutoff score used in Tauriel includes adjustment for age, while the FCSRT cutoff scores used in CREAD and CREAD2 do not, which potentially could have contributed to the different rates of screen failure on episodic memory criteria between Tauriel and CREAD2. However, in both studies, the mean age of participants who met the episodic memory criterion (Tauriel: 71.0, SD=6.9; CREAD2: 72.1, SD=7.6) was significantly higher than participants who failed this criterion (Tauriel: 69.9; SD=6.8; CREAD2: 70.2, SD=8.21; ps<0.05), which suggests that differences in age adjustment for cutoff scores is unlikely to explain our results. CREAD and Tauriel participants in the longitudinal mITT analysis cohorts were closely, but not completely matched. In particular, a higher percentage of CREAD participants met criteria for mild AD, and correspondingly, slightly lower baseline cognitive performance was seen in CREAD relative to Tauriel. However, our MMRM modeling explicitly adjusted for those variables to limit the impact of those differences on our results. Rater training and supervision for clinical outcome assessments were conducted by separate service providers across studies, raising the theoretical possibility that subtle systemic scoring biases could have obscured differences in rates of clinical progression. However, this seems unlikely, given that the administration and scoring of the assessments in question (CDR-SB, ADAS-Cog13, ADCS-ADL) are highly standardized and that raters at sites performing multiple AD clinical trials are frequently contemporaneously trained and monitored by multiple service providers.
Given that the FCSRT and RBANS produce similar rates of Aβ positivity and clinical progression, other considerations beyond these factors may drive the decision of what assessment to use for screening procedures in early AD clinical trials. These may include participant and staff burden, licensing costs for the assessments, and the potential utility of the FCSRT or RBANS as longitudinal outcome measures. Eligibility rates were lower when using the more stringent cutoffs for the FCSRT that were incorporated into CREAD and CREAD2, raising the possibility that screening cutoffs with this measure may require further customization to the disease stage under study. Further comparisons of the FCSRT and/or RBANS versus other episodic memory tests that have been used as inclusion criteria for early AD trials may provide additional guidance regarding which assessments can maximize screening efficiency and rates of disease progression in this patient population.
Funding: This work was supported by Genentech, Inc. and F. Hoffmann-La-Roche Ltd. Employees from both companies participated in design and conduct of the study; collection, management, analysis, and interpretation of the data, preparation, review, or approval of the manuscript, and decision to submit the manuscript for publication.
Acknowledgements: We would like to thank the all of the study participants and their families, and all of the site investigators, study coordinators, and staff. We would also like to thank Yihao Li for performing contributory statistical analyses. Assistance in preparing this article for publication was provided by Genentech, Inc.
Ethical standards: The study protocols were approved by Institutional Review Boards prior to patient recruitment and conducted in accordance with the International Conference on Harmonization E6 Guidelines for Good Clinical Practice. The study was carried out according to the laws of the country in which the trial was conducted. Each patient provided signed informed consent prior to study enrollment. All studies were registered at Clinicaltrials.gov: CREAD (NCT02670083), CREAD2 (NCT03114657), and Tauriel (NCT03289143).
Conflict of interest: ET, PTM, MS, KP, NH, SD, HS, MB, and KMS are current or former employees of Genentech, Inc., and shareholders of F. Hoffmann-La-Roche Ltd. GF and SO are full-time employees and shareholders of F. Hoffmann-La-Roche Ltd.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. Sperling RA, Jack CR, Jr., Aisen PS. Testing the right target and right drug at the right stage. Sci Transl Med 2011;3:111cm33. https://doi.org/10.1126/scitranslmed.3002609
2. Schneider LS. The potential and limits for clinical trials for early Alzheimer’s disease and some recommendations. J Nutr Health Aging 2010;14:295-8. https://doi.org/10.1007/s12603-010-0066-1
3. Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol 2014;13:614-29. https://doi.org/10.1016/S1474-4422(14)70090-0
4. Landau SM, Horng A, Fero A, Jagust WJ, Alzheimer’s Disease Neuroimaging Initiative. Amyloid negativity in patients with clinically diagnosed Alzheimer disease and MCI. Neurology 2016;86:1377-1385. https://doi.org/10.1212/WNL.0000000000002576
5. Tomadesso C, de La Sayette V, de Flores R, et al. Neuropsychology and neuroimaging profiles of amyloid-positive versus amyloid-negative amnestic mild cognitive impairment patients. Alzheimers Dement (Amst) 2018;10:269-277. https://doi.org/10.1016/j.dadm.2018.02.008
6. Macklin EA, Blacker D, Hyman BT, Betensky RA. Improved design of prodromal Alzheimer’s disease trials through cohort enrichment and surrogate endpoints. J Alzheimers Dis 2013;36:475-86. https://doi.org/10.3233/JAD-122212
7. Vuoksimaa E, McEvoy LK, Holland D, et al. Modifying the minimum criteria for diagnosing amnestic MCI to improve prediction of brain atrophy and progression to Alzheimer’s disease. Brain Imaging Behav 2020;14:787-796. https://doi.org/10.1007/s11682-018-0019-6
8. Grober E, Buschke H. Genuine memory deficits in dementia. Dev Neuropsychol 1987;3:13-36.
9. Randolph C, Tierney MC, Mohr E, Chase TN. The Repeatable Battery for the Assessment of Neuropsychological Status (RBANS): Preliminary clinical validity. J Clin Exp Neuropsychol 1998;20:310-9. https://doi.org/10.1076/jcen.20.3.310.823
10. Wagner M, Wolf S, Reischies FM, et al. Biomarker validation of a cued recall memory deficit in prodromal Alzheimer disease. Neurology 2012;78:379-86. https://doi.org/10.1212/WNL.0b013e318245f447
11. Teichmann M, Epelbaum S, Samri D, et al. Free and Cued Selective Reminding Test – accuracy for the differential diagnosis of Alzheimer’s and neurodegenerative diseases: A large-scale biomarker-characterized monocenter cohort study (ClinAD). Alzheimers Dement 2017;13:913-923. https://doi.org/10.1016/j.jalz.2016.12.014
12. Randolph C. The Utility of Episodic Memory Cut-Off Scores for Inclusion in Clinical Trials for Early Symptomatic Alzheimer Disease. Am J Geriatr Psychiatry 2019;27:1428-1432. https://doi.org/10.1016/j.jagp.2019.08.012
13. Sink K, Djakovic S, Smith JW, et al. FCSRT inclusion criteria support recruitment of a population with early Alzheimer’s disease likely to progress over 24 months: Results from the CREAD trial J Prev Alzheimers Dis 2019;6(Suppl 1):S19-S20.
14. Spallazzi M, Michelini G, Barocco F, et al. The role of free and cued selective reminding test in predicting [18F]Florbetaben PET results in mild cognitive impairment and mild dementia. J Alzheimers Dis 2020;73:1647-1659. https://doi.org/10.3233/JAD-190950
15. Suhrie KR, Hammers DB, Porter SM, et al. Predicting biomarkers in intact older adults and those with amnestic Mild Cognitive Impairment, and mild Alzheimer’s Disease using the Repeatable Battery for the Assessment of Neuropsychological Status. J Clin Exp Neuropsychol 2021;43:861-878. https://doi.org/10.1080/13803395.2021.2023476
16. Ostrowitski S, Bittner T, Sink KM, et al. Evaluating the safety and efficacy of crenezumab vs placebo in adults with early Alzheimer disease: Two Phase 3 randomized placebo-controlled trials. JAMA Neurol 2022;https://doi.org/10.1001/jamaneurol.2022.2909
17. Teng E, Manser PT, Pickthorn K, et al. Safety and efficacy of semorinemab in individuals with prodromal to mild Alzheimer’s disease: A randomized clinical trial. JAMA Neurol 2022;https://doi.org/10.1001/jamaneurol.2022.1375
18. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7:270-9. https://doi.org/10.1016/j.jalz.2011.03.008
19. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7:263-9. https://doi.org/10.1016/j.jalz.2011.03.005
20. Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry 1982;140:566-72. https://doi.org/10.1192/bjp.140.6.566
21. Imaging P. Neuraceq (florbetaben F 18 injection) [package insert]. U.S. Food and Drug Administration website. Updated 3/2014. Accessed 12/28/2020, https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204677s000lbl.pdf
22. Healthcare G. Vizamyl (flutemetamol F 18 injection) [package insert]. U.S. Food and Drug Administration website. Updated 4/2016. Accessed 12/28/2020, https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/203137s005lbl.pdf
23. Lilly E. Amyvid (florbetapir F 18 injection) [package insert]. U.S. Food and Drug Administration website. Updated 4/2012. Accessed 12/29/2020, https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202008s000lbl.pdf
24. Ng KP, Pascoal TA, Mathotaarachchi S, et al. Monoamine oxidase B inhibitor, selegiline, reduces (18)F-THK5351 uptake in the human brain. Alzheimers Res Ther 2017;9:25. https://doi.org/10.1186/s13195-017-0253-y
25. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189-98. https://doi.org/10.1016/0022-3956(75)90026-6
26. Smith J, Edgar CJ, Lasser RA, et al. Free and Cued Selective Reminding Test as an inclusion criterion for early Alzheimer’s disease clinical trial populations. J Prev Alzheimers Dis 2016;3:P343-P344.
27. Grober E, Sanders AE, Hall C, Lipton RB. Free and cued selective reminding identifies very mild dementia in primary care. Alzheimer Dis Assoc Disord 2010;24:284-90. https://doi.org/10.1097/WAD.0b013e3181cfc78b
28. Randolph C. Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) Manual. The Psychological Corporation; 1998.
29. McDougall F, Edgar C, Mertes M, et al. Psychometric properties of the clinical dementia rating – sum of boxes and other cognitive and functional outcomes in a prodromal Alzheimer’s disease population. J Prev Alzheimers Dis 2021;8:151-160. https://doi.org/10.14283/jpad.2020.73
30. Duff K, Beglinger LJ, Schoenberg MR, et al. Test-retest stability and practice effects of the RBANS in a community dwelling elderly sample. J Clin Exp Neuropsychol 2005;27:565-75. https://doi.org/10.1080/13803390490918363
31. Mohs RC, Knopman D, Petersen RC, et al. Development of cognitive instruments for use in clinical trials of antidementia drugs: additions to the Alzheimer’s Disease Assessment Scale that broaden its scope. The Alzheimer’s Disease Cooperative Study. Alzheimer Dis Assoc Disord 1997;11 Suppl 2:S13-21.
32. Galasko D, Bennett D, Sano M, et al. An inventory to assess activities of daily living for clinical trials in Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. Alzheimer Dis Assoc Disord 1997;11 Suppl 2:S33-9.
33. Team RC. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2018. Accessed 1 Sep 2020. http://www.R-project.org/.
34. Girtler N, De Carli F, Amore M, et al. A normative study of the Italian printed word version of the Free and Cued Selective Reminding Test. Neurol Sci 2015;36:1127-34. https://doi.org/10.1007/s10072-015-2237-7
35. Negash S, Weber C, Randolph C. Clinical and amyloid screen failure rates in episodic memory measures in early AD trials. J Prev Alzheimers Dis 2018;5:S012.
36. Petersen RC, Aisen PS, Beckett LA, et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology 2010;74:201-9. https://doi.org/10.1212/WNL.0b013e3181cb3e25
37. Tosun D, Veitch D, Aisen P, et al. Detection of beta-amyloid positivity in Alzheimer’s Disease Neuroimaging Initiative participants with demographics, cognition, MRI and plasma biomarkers. Brain Commun 2021;3:fcab008. https://doi.org/10.1093/braincomms/fcab008
38. Pike KE, Savage G, Villemagne VL, et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain 2007;130:2837-44. https://doi.org/10.1093/brain/awm238
39. Sarazin M, Berr C, De Rotrou J, et al. Amnestic syndrome of the medial temporal type identifies prodromal AD: A longitudinal study. Neurology 2007;69:1859-67. https://doi.org/10.1212/01.wnl.0000279336.36610.f7
40. Doecke JD, Ward L, Burnham SC, et al. Elecsys CSF biomarker immunoassays demonstrate concordance with amyloid-PET imaging. Alzheimers Res Ther 2020;12:36. https://doi.org/10.1186/s13195-020-00595-5
41. Palmqvist S, Mattsson N, Hansson O, Alzheimer’s Disease Neuroimaging Initiative. Cerebrospinal fluid analysis detects cerebral amyloid-beta accumulation earlier than positron emission tomography. Brain 2016;139:1226-36. https://doi.org/10.1093/brain/aww015
42. Florian H, Wang D, Guo Q, et al. Phase 2 study of tilavonemab, an anti-tau antibody, in early Alzheimer’s disease. J Prev Alzheimers Dis 2021;8:S50-S51.
43. Swanson CJ, Zhang Y, Dhadda S, et al. A randomized, double-blind, Phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Abeta protofibril antibody. Alzheimers Res Ther 2021;13:80. https://doi.org/10.1186/s13195-021-00813-8
44. Shulman M, Rajagovindan R, Kong J, et al. Top-line results from TANGO, a Phase 2 study of gosuranemab in participants with mild cognitive impairment due to Alzheimer’s disease and mild Alzheimer’s disease. J Prev Alzheimers Dis 2021;8:S65-S66.
© The Authors 2022