
S. Budd Haeberlein1, P.S. Aisen2, F. Barkhof3,4, S. Chalkias1,*, T. Chen1, S. Cohen5, G. Dent1, O. Hansson6,7, K. Harrison1, C. von Hehn1,*, T. Iwatsubo8, C. Mallinckrodt1,*, C.J. Mummery9, K.K. Muralidharan1, I. Nestorov1, L. Nisenbaum1,*, R. Rajagovindan1,*, L. Skordos1,*, Y. Tian1, C.H. van Dyck10, B. Vellas11, S. Wu1, Y. Zhu1, A. Sandrock1,*
1. Biogen, Cambridge, MA, USA; 2. Alzheimer’s Therapeutic Research Institute, University of Southern California, San Diego, CA, USA; 3. Department of Radiology and Nuclear Medicine, Amsterdam UMC, Amsterdam, the Netherlands; 4. UCL Queen Square Institute of Neurology & Centre for Medical Image Computing, London, UK; 5. Toronto Memory Program, Toronto, ON, Canada; 6. Clinical Memory Research Unit, Department of Clinical Sciences, Lund University, Malmö, Sweden; 7. Memory Clinic, Skåne University Hospital, Malmö, Sweden; 8. Department of Neuropathology, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan; 9. Dementia Research Centre, Queen Square Institute of Neurology, University College London, London, UK; 10. Alzheimer’s Disease Research Unit, Yale School of Medicine, New Haven, CT, USA; 11. Toulouse Gerontopole University Hospital, Universite Paul Sabatier, INSERM U 1295, France; *Denotes an author who was an employee of Biogen at the time of this study and who has since left the company.
Corresponding Author: Samantha Budd Haeberlein, Biogen, Cambridge, Massachusetts, 617-679-3159, samantha.buddhaeberlein@biogen.com
J Prev Alz Dis 2022;
Published online March 18, 2022, http://dx.doi.org/10.14283/jpad.2022.30
Abstract
BACKGROUND: Alzheimer’s disease is a progressive, irreversible, and fatal disease for which accumulation of amyloid beta is thought to play a key role in pathogenesis. Aducanumab is a human monoclonal antibody directed against aggregated soluble and insoluble forms of amyloid beta.
OBJECTIVES: We evaluated the efficacy and safety of aducanumab in early Alzheimer’s disease.
DESIGN: EMERGE and ENGAGE were two randomized, double-blind, placebo-controlled, global, phase 3 studies of aducanumab in patients with early Alzheimer’s disease.
SETTING: These studies involved 348 sites in 20 countries.
PARTICIPANTS: Participants included 1638 (EMERGE) and 1647 (ENGAGE) patients (aged 50–85 years, confirmed amyloid pathology) who met clinical criteria for mild cognitive impairment due to Alzheimer’s disease or mild Alzheimer’s disease dementia, of which 1812 (55.2%) completed the study.
INTERVENTION: Participants were randomly assigned 1:1:1 to receive aducanumab low dose (3 or 6 mg/kg target dose), high dose (10 mg/kg target dose), or placebo via IV infusion once every 4 weeks over 76 weeks.
MEASUREMENTS: The primary outcome measure was change from baseline to week 78 on the Clinical Dementia Rating Sum of Boxes (CDR-SB), an integrated scale that assesses both function and cognition. Other measures included safety assessments; secondary and tertiary clinical outcomes that assessed cognition, function, and behavior; and biomarker endpoints.
RESULTS: EMERGE and ENGAGE were halted based on futility analysis of data pooled from the first approximately 50% of enrolled patients; subsequent efficacy analyses included data from a larger data set collected up to futility declaration and followed prespecified statistical analyses. The primary endpoint was met in EMERGE (difference of −0.39 for high-dose aducanumab vs placebo [95% CI, −0.69 to −0.09; P=.012; 22% decrease]) but not in ENGAGE (difference of 0.03, [95% CI, −0.26 to 0.33; P=.833; 2% increase]). Results of biomarker substudies confirmed target engagement and dose-dependent reduction in markers of Alzheimer’s disease pathophysiology. The most common adverse event was amyloid-related imaging abnormalities-edema.
CONCLUSIONS: Data from EMERGE demonstrated a statistically significant change across all four primary and secondary clinical endpoints. ENGAGE did not meet its primary or secondary endpoints. A dose- and time-dependent reduction in pathophysiological markers of Alzheimer’s disease was observed in both trials.
Key words: Aducanumab, Alzheimer’s disease, amyloid beta.
Introduction
Alzheimer’s disease (AD) is a progressive, irreversible, and fatal neurological disorder. Accumulation of amyloid beta (Aβ) species in the brain is a primary pathological feature of Alzheimer’s disease and can occur decades prior to the onset of clinical symptoms. While Aβ is known to exert neuronal toxicity, it is also postulated to be responsible for downstream pathologies, such as tau phosphorylation and aggregation, that lead to neuronal death in AD (1-3).
Targeting the amyloid cascade has been a key focus for many clinical development programs in AD over the last 25 years (3, 4). These efforts have been reviewed extensively (3). Earlier phase 3 trials of investigational anti-Aβ monoclonal antibodies did not demonstrate efficacy and failed to reduce Aβ plaque levels in a clinical trial setting (5-7). These studies also recruited patients at later stages of disease and included individuals without evidence of Aβ pathology (i.e., patients without Alzheimer’s disease). In contrast, emerging data from second-generation anti-Aβ antibodies demonstrates a robust reduction in the levels of Aβ plaques in patients in the earlier stages of AD (8-10).
Aducanumab is a human monoclonal antibody that selectively targets aggregated forms of Aβ, including soluble oligomers and insoluble fibrils (8). In a prior phase 1b study (PRIME), aducanumab treatment resulted in dose- and time-dependent reduction in Aβ plaques, accompanied by slowed clinical decline (exploratory endpoint) (8).
Two identically designed phase 3 trials, EMERGE and ENGAGE, assessed the efficacy and safety of aducanumab in patients with early AD (mild cognitive impairment [MCI] due to AD and mild AD dementia). Both trials were halted early based on results from a futility analysis of interim data. Although the prespecified futility criteria were met, suggesting treatment was unlikely to demonstrate clinical benefit, it was later determined that two assumptions on which the futility analysis was based were violated. These assumptions were 1) that the treatment effect in the two studies would be similar and 2) constancy of effect (i.e., that later enrolled patients would have the same effect as earlier enrolled patients. Therefore, results of the futility analysis yielded inaccurate predictions for the final outcomes. Data collected per protocol and under double- blind conditions up until futility announcement were subsequently analyzed based on the prespecified analysis plan.
Here, we describe the primary efficacy and safety results from these studies, and findings from the biomarker substudies.
Methods
Clinical study patients
EMERGE (NCT02484547) and ENGAGE (NCT02477800) included patients aged 50 to 85 years who met clinical criteria for MCI due to AD (13) or mild AD dementia (14), with amyloid pathology confirmed by visual assessment of amyloid positron emission tomography (PET; 18F-florbetapir, 18F-flutemetamol, or 18F-florbetaben). This patient population is consistent with stage 3 and 4 patients as described in the FDA 2018 Guidance for Industry Early Alzheimer’s Disease: Developing Drugs for Treatment (15). Among the inclusion criteria (Supplement 1) were a Mini-Mental State Examination (MMSE) (16) score of 24 to 30 and a Clinical Dementia Rating Scale (CDR) (17) global score of 0.5. Brain magnetic resonance imaging (MRI) was used to exclude patients with confounding pathologies, including acute or sub-acute hemorrhage, more than four microhemorrhages, cortical infarcts, >1 lacunar infarct, superficial siderosis, or a history of white matter disease as defined per protocol, or conditions that posed a risk to the patient or prevented MRI monitoring (full exclusion criteria are listed in Supplement 1). Patients with medical conditions possibly contributing to cognitive impairment were also excluded. Stable use of concomitant medications for chronic conditions was permitted during the study, except as defined in the protocol. Use of aspirin at a prophylactic dose (≤325 mg daily) was permitted, but use of any other medications with anti-platelet or anti-coagulant properties was exclusionary. For cholinesterase inhibitors and memantine, patients were required to be on a stable dose before screening, with no dose adjustment during the study. Vaccinations with live or attenuated vaccines were allowed during the study.
Clinical study design
EMERGE and ENGAGE were randomized, double-blind, placebo-controlled trials conducted at 348 sites in 20 countries. The number of patients enrolled in ENGAGE remained ahead of EMERGE throughout the study enrollment period (Supplemental Data Fig. 1a). Patients were randomized (1:1:1) to receive low-dose aducanumab, high-dose aducanumab, or placebo (Supplemental Data Fig. 1b) via intravenous infusion following dilution into saline every 4 weeks over 76 weeks (20 doses total). The randomization was stratified by site and apolipoprotein E (ApoE) ε4 carrier status. The dose in the low-dose group was titrated to a target dose of 3 mg/kg (ApoE ε4+) or 6 mg/kg (ApoE ε4−). The dose in the high-dose group was titrated to a target dose of 6 mg/kg (ApoE ε4+) or 10 mg/kg (ApoE ε4−) prior to protocol amendments. Based on findings from a prior study with aducanumab (PRIME) (8), two protocol amendments were implemented that aimed to enable more participants in the high-dose arms to achieve the target dose of 10 mg/kg. In protocol version 3 (PV3; approved July 21, 2016), participants who suspended dosing due to amyloid-related imaging abnormalities (ARIA) could, after resolution of ARIA, resume dosing at the same dose and continue titration to the target dose (rather than resume at the next lower dose, with no further increases in dose permitted). This protocol amendment applied to all participants who consented. To maximize the dose-dependent effect of aducanumab (8), the target dose for ApoE ε4+ carriers in the high-dose regimen was increased from 6 to 10 mg/kg in protocol version 4 (PV4; approved March 24, 2017, and implemented over approximately 18 months across sites). Of note, although all patients were also asked for consent to PV4, the amendment had changes within it that impacted selectively the ApoE ε4 carriers. Approximately two-thirds of the trial participants were ApoE ε4+ carriers. While each of these amendments impacted the opportunities for receiving 10 mg/kg, PV4 had the greater potential to impact a larger number of patients in the trial. Due to differences in the rates of enrollment, these two protocol amendments influenced more patients in EMERGE than ENGAGE because EMERGE started later and enrolled more patients after each amendment (Supplemental Data Fig. 1a). At the time of the PV4 amendment, ENGAGE had enrolled approximately 200 more participants than EMERGE (Supplemental Data Fig. 1a).
Safety monitoring
Safety monitoring comprised reports of adverse events, including ARIA; assessment of vital signs; physical and neurological examinations; electrocardiography; hematologic and serum chemical testing; urinalysis; and brain MRI scans reviewed locally and by a central radiologist with expertise in ARIA. ARIA can manifest as brain edema or sulcal effusion (ARIA-E) or as hemosiderin deposits in the brain parenchyma (ARIA-H microhemorrhage) or on the pial surface (ARIA-H superficial siderosis) (18), with each reported instance classified as such in the trials. The presence of vasogenic edema or effusion was evaluated using T2 fluid-attenuated inversion recovery (FLAIR) as the primary diagnostic imaging sequence (18). ARIA-E severity was classified based on the number and size of any edematous regions. A single region <5 cm was considered of mild radiographic severity, a single region 5 to 10 cm or multiple regions all <10 cm were considered moderate, and any region >10 cm was considered severe. The presence of any new incident brain microhemorrhage or localized superficial siderosis events was also evaluated using gradient echo sequences (GRE). Greater or equal to 1 and ≤4 new incident microhemorrhages were considered mild, ≥5 and ≤9 moderate, and ≥10 severe. One new incident area of superficial siderosis was considered mild, 2 moderate, and >2 severe.
Brain MRI was conducted at screening and in weeks 14, 22, 30, 42, 54, 66, and 78. All brain MRI findings of ARIA were required to be reported as adverse events, even in the absence of clinical symptoms. For radiographically mild ARIA that were clinically asymptomatic, dosing continued without interruption; if symptoms were present, dosing was suspended. Detection of moderate or severe ARIA-E or of moderate ARIA-H led to dosing suspension until resolution of ARIA-E or stabilization of ARIA-H. Symptomatic ARIA-H (microhemorrhages or superficial siderosis) with serious clinical symptoms were required to permanently discontinue study treatment (Supplement 2). Radiographically severe ARIA-H or brain hemorrhage of >1 cm in diameter resulted in discontinuation of dosing permanently. Detection of an ARIA episode was followed by MRI scans conducted approximately every 4 weeks to document resolution of ARIA-E or stabilization of ARIA-H. The investigator was responsible for evaluating patients for any symptoms observed in the setting of ARIA.
Clinical assessments and biomarker substudies
The primary outcome measure was the CDR-sum of boxes (CDR-SB) (17), an assessment of both cognition and function in AD. Secondary clinical outcome measures also assessed cognitive decline (MMSE [16] and Alzheimer’s Disease Assessment Scale–Cognitive Subscale–13 items [ADAS-Cog13] [19]) and ability to perform daily activities (Alzheimer’s Disease Cooperative Study Activities of Daily Living Inventory–Mild Cognitive Impairment [ADCS-ADL-MCI]) (20). The Neuropsychiatric Inventory-10 (NPI-10) (21), a caregiver-based assessment of neuropsychiatric symptoms, was the lone tertiary efficacy endpoint. These clinical outcome measures were assessed during screening and at weeks 26, 50, and 78. Three health care professionals were required for each efficacy visit: 1) the treating physician was responsible for the management of ARIA, routine neurological care, and assessment and treatment of adverse events; PIs could not serve as rating HCPs and did not have access to the post-baseline efficacy data; 2) an independent rater administered the primary efficacy endpoint; 3) a second independent rater administered the secondary efficacy endpoints. The two independent raters were not involved in participant care and management and were blinded to ARIA and other medical information to minimize the potential for functional unblinding. Unblinded pharmacy staff managed study treatment receipt, dispensing, and preparation. Treatment assignments were not shared with the participants, their families, or any member of the blinded study team. To ensure consistency across sites, efficacy raters completed the standardized study-specific qualification process on clinical efficacy assessment scoring, and all sites attempted to maintain the same raters throughout the study for specific assessments and for each participant. A qualified approved back-up rater conducted the assessments only in extenuating circumstances resulting in unavailability.
Longitudinal amyloid PET imaging using 18F-florbetapir was performed in a subset of patients (n=488 in EMERGE; n=585 in ENGAGE) at screening, week 26, and week 78. The cortical composite standardized uptake value ratio (SUVR) was derived according to methods previously described and further detailed in Supplement 3 (8, 22). The composite SUVR was also transformed to centiloid (CL) units (23).
Longitudinal tau PET imaging using 18F-MK-6240 was performed in a subset of patients (n=37, pooled across studies) at both screening and week 78. SUVR composite regions and normalization are detailed in Supplement 3. Due to early termination of the studies, the median postbaseline visit occurred at 13.6 months (range, 9.5 to 19.6 months).
Cerebrospinal fluid (CSF) was collected at both baseline and week 78 in a subset of patients (n=78 in EMERGE; n=53 in ENGAGE). CSF levels of Aβ1-42, phosphorylated tau181 (p-tau), and total tau (t-tau) were measured using the Lumipulse G immunoassays (Fujirebio).
Lumbar puncture was used to collect CSF samples from living clinical trial CSF substudy participants with early AD via a 22g Sprotte atraumatic needle inserted between the L3/L4 or L4/L5 interspace. Time of collection was recorded. Samples were collected at room temperature following usual and customary sterile techniques and stored in polypropylene tubes at −70°C for 7 to 52 months until analysis.
At week 78, the effects of aducanumab treatment on plasma-tau181 levels were assessed in clinical trial participants with early AD. A 6-mL tube of whole blood was collected using a K2EDTA tube and processed according to standard procedures with centrifugation at room temperature to separate cells and plasma within 1 hour of sample collection. Following centrifugation, samples were aliquoted into 2-mL polypropylene tubes and frozen immediately at ‒70°C until shipment (except where unavailable, in which case samples were stored at -20°C). Samples were stored buffer-free between -70°C and -80°C until analysis for approximately 6 years. Only the intent-to-treat (ITT) patients with plasma samples available at both screening and week 78 were selected for further analyses. Available samples at screening, week 56 (week 48 if under PV1–3), and week 78 were tested. A total of 6684 plasma samples (n=3474 from EMERGE and n=3210 from ENGAGE) were analyzed using the Quanterix Simoa p-tau181 Advantage V2 kit at Frontage Laboratories’ (Exton, PA) CLIA laboratory. Data were captured by the Quanterix Simoa HD-X Analyzer. Watson LIMS Version 7.6 was used for data regression. The standard curve was fitted with a four-parameter logistic (Marquardt) regression model with a weighting factor of 1/Y2. Concentration was presented in pg/mL, and coefficient of variation (CV) and relative error (RE) as percentages. The inter-assay CV was 6.49–8.15%, and the intra-assay CV was 8.30–9.21%.
Statistical analyses
A sample size of 450 per study treatment group (1350 total per study) was calculated based on a 90% power to detect a mean difference of 0.5 in change from baseline in CDR-SB score at week 78, based on a two-sided .05 test, assuming an SD of 1.92 and a 30% dropout rate. An assumed true mean difference of 0.5 between the two treatment groups would represent an approximately 25% reduction in the placebo mean change from baseline at week 78, using an estimated placebo mean change of 2.0 on the CDR-SB score. As prespecified in the protocol, sample size was reassessed based on assessment of variance approximately 3 months before enrollment completion. Sample size was adjusted to 535 per arm to maintain 90% power with the observed higher than anticipated variance. Data analysis was performed by SW and TC.
The primary analysis assessed the mean difference between treatments in change from baseline CDR-SB score at week 78 in the randomized and dosed population (all randomized patients who had received ≥1 dose of study treatment). Data collected on or after the futility declaration on March 21, 2019, were excluded to minimize the potential for bias introduced by the futility declaration. A mixed model for repeated measures (MMRM) was used to assess CDR-SB, MMSE, ADAS-Cog13, ADCS-ADL-MCI, and NPI-10, with fixed effects of treatment, categorical visit, treatment-by-visit interaction, baseline score, baseline score-by-visit interaction, baseline MMSE score (same as baseline score in the MMSE model), AD symptomatic medication use at baseline, region, and ApoE ε4 status (carrier and noncarrier). Other than the primary analysis for the primary and secondary endpoints, P values were all nominal.
Analyses related to biomarkers were performed in a subset of participants in each study (baseline disease characteristics for substudies are provided as Supplemental Data Table 1), due to data availability (e.g., biomarkers were not collected in all patients, per protocol). The safety MRI population consisted of fewer participants than that of the randomized and dosed population, as this requires a post-baseline MRI.
The primary analysis of the efficacy endpoints assumed that missing data were missing at random. Different assumptions for missing data were explored as part of prespecified sensitivity analyses (Supplement 4). A sequential testing procedure, prespecified in the study protocols, was used to control the type I error rate due to multiple endpoints and multiple comparisons. The clinical principle underpinning the testing strategy was that high dose (10 mg/kg) was the target dose. Therefore, failure of the low dose at any endpoint should not preclude testing of the high dose. The multiplicity testing procedure for the primary and secondary endpoints was as follows:
• Sequential testing was rank prioritized: CDR-SB, MMSE, ADAS-Cog13, ADCS-ADL-MCI
• If the high dose was significant for endpoint x, test the high dose for endpoint x+1 and low dose for endpoint x
• If the low dose was not significant for endpoint x, all endpoints of lower rank for low dose are not significant
• If the high dose was not significant for endpoint x, endpoint x for low dose and all endpoints of lower rank for the high and low doses are not significant.
As part of the sensitivity analysis of the primary endpoint, data were analyzed for normality, and non-normally distributed variables were transformed or analyzed with appropriate nonparametric tests, as described in the legend of Supplemental Data Table 2.
Change from baseline in amyloid PET composite SUVR was analyzed using an MMRM with fixed effects of treatment, categorical visit, treatment-by-visit interaction, baseline SUVR, baseline SUVR-by-visit interaction, baseline MMSE score, ApoE ε4 status (carrier and noncarrier), and baseline age.
MMRM analyses were also conducted to assess the effect of aducanumab on change from baseline in plasma p-tau181 levels (using data from the placebo-controlled period; fixed effects included visit, treatment group and its interaction with visit, baseline value and its interaction with visit, age, and ApoE ε4 status. Correlation analyses were conducted to assess the following: 1) relationship between change from baseline in plasma p-tau181 levels and amyloid PET composite SUVR (assessed in three pooled treatment arms); 2) relationship between change from baseline in plasma p-tau181 levels and clinical decline (assessed in pooled low- and high-dose arms). Data were presented using partial Spearman correlation coefficients adjusting for baseline plasma p-tau181 levels, baseline amyloid PET SUVR (for 1) or baseline clinical scores (for 2), and age.
Tau PET SUVR and CSF biomarkers were analyzed using analysis of covariance models (Supplement 3).
All biomarker analyses were exploratory; amyloid PET was the only biomarker outcome for which a sample size calculation was performed.
All statistical tests were two-sided tests. The statistical software, SAS®, was used for all summaries and analyses.
Futility analysis
An interim analysis for futility was prespecified in the study protocols and statistical analysis plan to allow for early termination of the studies in the event that the analysis predicted the drug to be ineffective. To maintain the integrity of the study, blinded data were provided to an external vendor (IQVIA). A group of statisticians and programmers at IQVIA conducted the unblinded analyses for the futility analysis. An independent data monitoring committee of experienced clinical (expert academic neurologists and AD clinical trialists) and statistical experts reviewed the analysis. Futility analysis methodology was pre-specified in the analysis table shells and specifications that were provided to the IQVIA team and the independent data monitoring committee for review in advance of the futility analysis.This interim analysis was performed, per protocol, after approximately 50% of the participants (whose data were used) had the opportunity to complete week 78. The prespecified criteria for futility were primarily based on conditional power for CDR-SB, which is the probability calculated on the data at the interim analysis that the final analysis would show statistical significance in favor of aducanumab. The studies were to be considered futile if the conditional power was < 20% for each arm of both studies. The conditional power for each study was calculated, with a future estimate calculated based on pooled data from EMERGE and ENGAGE. The use of pooled data for the future estimate was based on the assumption that the pooling approach had better operating characteristics than the approach based on single-trial data, in the event that small to moderate heterogeneity with regard to treatment effects existed between the two studies (24). As the two phase 3 studies were identically designed, large heterogeneity was not anticipated.
Two assumptions on which the futility analysis was based were violated: the assumption that the treatment effect would be similar in the two studies and that the treatment effect would not substantially change during the study). Therefore, results of the futility analysis yielded inaccurate predictions for the final outcomes.
Availability of data and materials
The data described in this article are not publicly available. The authors and Biogen are fully supportive of data sharing. Biogen has established processes to share protocols, clinical study reports, study-level data, and de-identified patient-level data. These data and materials will be made available to qualified scientific researchers to achieve the objective(s) in their approved, methodologically sound research proposal following US and EU marketing approval of aducanumab for the treatment of AD, with no end date. Proposals should be submitted through Vivli (https://vivli.org). To gain access, data requestors will need to sign a data sharing agreement. Data are made available for 1 year on a secure platform. For general inquiries, please contact datasharing@biogen.com. Biogen’s data-sharing policies and processes are detailed on the website http://clinicalresearch.biogen.com.
Results
Disposition and baseline characteristics
ENGAGE (first treatment: August 13, 2015) started 1 month earlier than EMERGE (first treatment: September 15, 2015); recruitment for both ended in July 2018. The randomized and dosed population (all randomized patients who received at least one dose of study treatment) included 1638 patients in EMERGE and 1647 in ENGAGE (Figure 1). No obvious imbalances in baseline demographics were noted across treatment arms (Table 1).
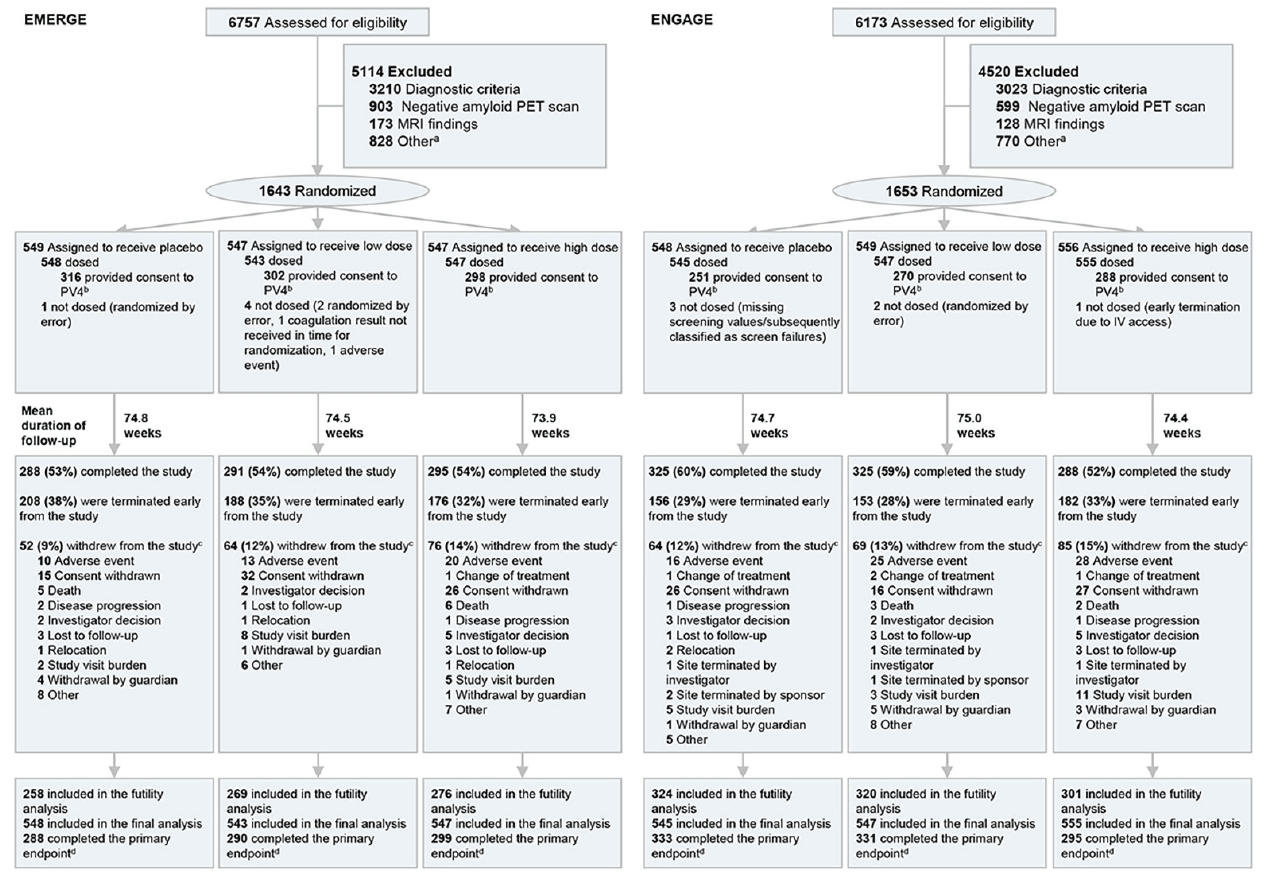
Figure 1. Patient disposition
Intent-to-treat population; a. Other reasons for not meeting inclusion/exclusion criteria include inability to adhere to study requirements; presence of diabetes that, in the judgment of the investigator, cannot be controlled or adequately managed; inability to understand the purpose and risks of the study and provide signed and dated informed consent and authorization to use protected health information in accordance with national and local privacy regulations; other unspecified reasons that, in the opinion of the investigator or sponsor, make the participant unsuitable for enrollment; history of or positive test result at screening for hepatitis C virus antibody or hepatitis B virus (defined as positive for both hepatitis B surface antigen and hepatitis B core antibody); use of allowed chronic medications at doses that have not been stable for ≥4 weeks prior to screening visit 1 and screening up to day 1, or use of medications for AD at doses that have not been stable for ≥ 8 weeks; and unknown/unclear; b. By week 16, which reflects the opportunity at which the patients can receive the full dose range; c. Some categories with <1% patients are not displayed, including loss of capacity, pregnancy, and protocol amendment; d. Completed the primary endpoint prior to futility declaration on March 21, 2019; IV, intravenous; PET, positron emission tomography.
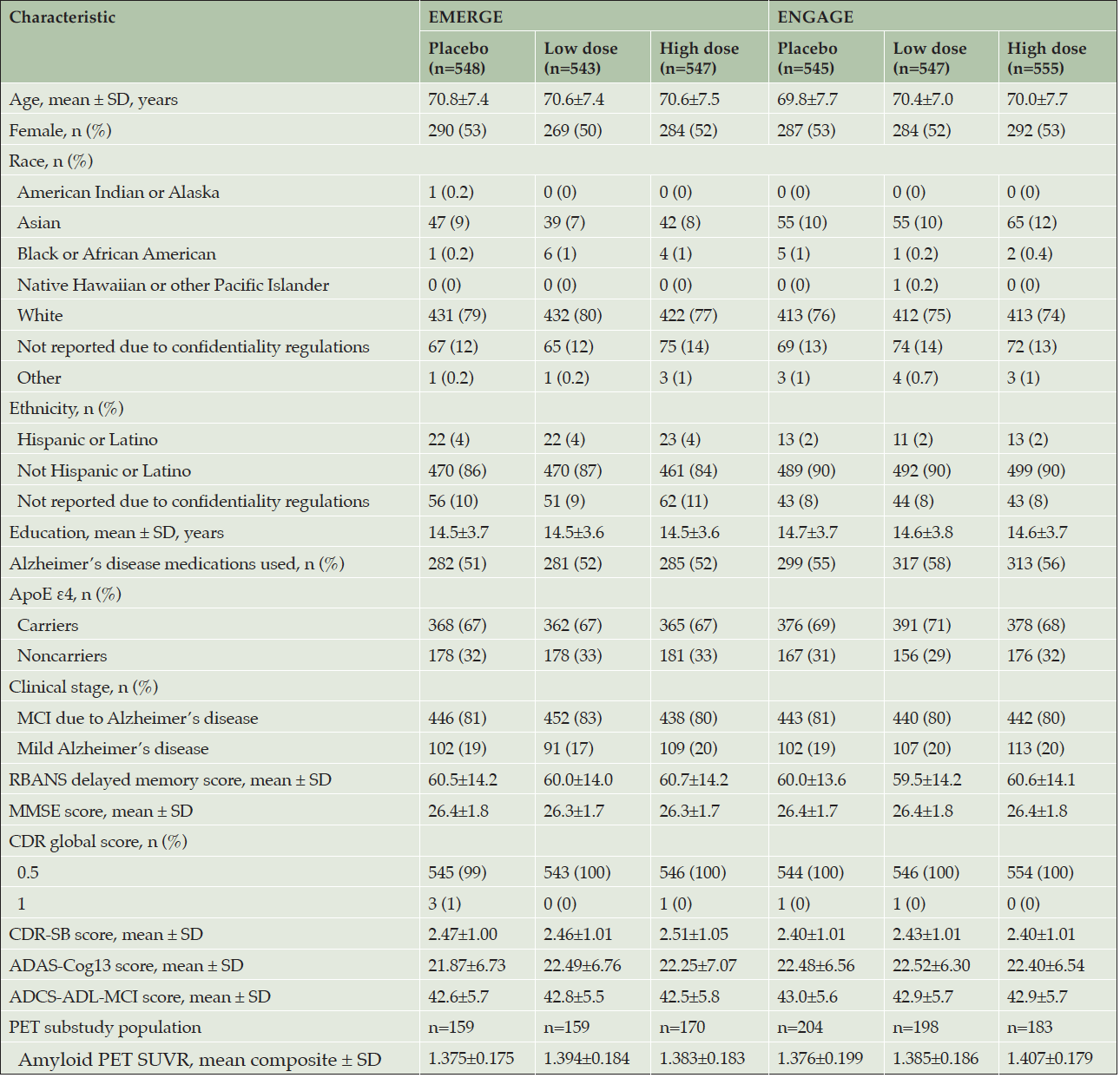
Table 1. Demographic and baseline disease characteristics
Randomized and dosed population; ADAS-Cog13, Alzheimer’s Disease Assessment Scale–cognitive subscale (13 items); ADCS-ADL-MCI, Alzheimer’s Disease Cooperative Study–Activities of Daily Living Inventory (mild cognitive impairment version); ApoE, apolipoprotein E; CDR-SB, Clinical Dementia Rating–sum of boxes; MCI, mild cognitive impairment; MMSE, Mini-Mental State Examination; PET, positron emission tomography; RBANS, Repeatable Battery for Assessment of Neuropsychological Status; SUVR, standardized uptake value ratio.
Futility analysis
Futility analysis included data from 49% patients in EMERGE and 57% patients in ENGAGE who had the opportunity to complete the week 78 visit by December 26, 2018. The prespecified futility criteria were met (see Methods section). An independent data monitoring committee reviewed the unblinded results of the interim analysis and made the recommendation to the sponsor to terminate the studies. Both studies were terminated on March 21, 2019. Following the futility announcement (March 21, 2019), all dosing stopped at the study sites, and data collection and data cleaning continued.
The prespecified futility methodology used pooled data from EMERGE and ENGAGE to predict the future unobserved treatment effect. However, the individual study results using the prespecified primary efficacy analysis methods on the futility data set showed a −18% treatment difference on the CDR-SB, favoring high-dose aducanumab in EMERGE, and a 15% treatment difference on CDR-SB, favoring placebo in ENGAGE. This outcome violated an assumption on which the estimation of conditional power was based—namely, that the treatment effect in the two studies would be similar. Consequently, conditional power was subsequently recalculated using the data from each of the two studies to predict the future unobserved treatment effect, and this nonpooled analysis yielded estimates of 59% and 0% on the primary endpoint for the high-dose groups in EMERGE and ENGAGE, respectively. With this analysis, the futility criteria would have not been met.
Final database lock was on November 13, 2019, for EMERGE and November 15, 2019, for ENGAGE. However, the final data set for primary efficacy analysis included efficacy data collected, under double-blind conditions, up to March 20, 2019—the day prior to the futility announcement, and this final data set included 65.2% more data observations than the futility data set (Supplemental Data Table 3). We present the final analyses for these studies.
Efficacy
The primary endpoint was met in EMERGE. High-dose aducanumab demonstrated a difference of −0.39 vs placebo in the mean change from baseline in CDR-SB score at week 78 (95% CI, −0.69 to −0.09; P=.012), a 22% reduction in decline (Table 2, Supplemental Data Fig. 2a).
The high-dose aducanumab arm also showed less decline vs placebo on each of the three prespecified secondary endpoints (Table 2, Supplemental Data Fig. 2b-d): MMSE (difference of 0.6 vs placebo in mean change from baseline; −18%; P=.049), ADAS-Cog13 (difference of −1.40 vs placebo in mean change from baseline; −27%; P=.010), and ADCS-ADL-MCI (difference of 1.7 vs placebo in mean change from baseline; −40%; P<.001) at week 78. Results from three out of four endpoints from the low-dose aducanumab arm were numerically intermediate between those of the high-dose and placebo arms but not statistically significant vs placebo on any primary or secondary endpoint (Table 2, Supplemental Data Fig. 2a-d). Change from baseline on the tertiary efficacy endpoint, NPI-10, at week 78 was −1.3 vs placebo in mean change from baseline with high-dose aducanumab (−87%; P=.022) (Supplemental Data Fig. 2e). Sensitivity analyses were conducted, and each confirmed the statistically significant high-dose aducanumab results in EMERGE (Supplemental Data Table 2).
High-dose aducanumab showed a numerical advantage over placebo in all prespecified subgroups for the primary endpoint, and 47 of 48 prespecified subgroups for the secondary endpoints in EMERGE (Supplemental Data Fig. 3a-d). Low-dose aducanumab results are presented in Supplemental Data Fig. 3i-l.
The primary endpoint was not met in ENGAGE. The difference in mean change from baseline in CDR-SB scores at week 78 was 0.03 with high-dose aducanumab vs placebo (95% CI, −0.26 to 0.33; P=.833), an increase of 2% (Table 2, Supplemental Data Fig. 2a). In the high-dose aducanumab arm, the three prespecified secondary endpoints were also not met. The differences in mean change from baseline at week 78 vs placebo on the MMSE (−0.1; 3%; P=.811), ADAS-Cog13 (−0.59; −11%; P=.258), ADCS-ADL-MCI (0.7; −18%; P=.151), and NPI-10 (0.1; 8%; P=.907) (Supplemental Data Fig. 2b-e) were also not statistically significant. Results from the low-dose aducanumab arm were not statistically significant vs placebo on any primary or secondary endpoint (Table 2, Supplemental Data Fig. 2a-d), and were consistent with those from EMERGE.
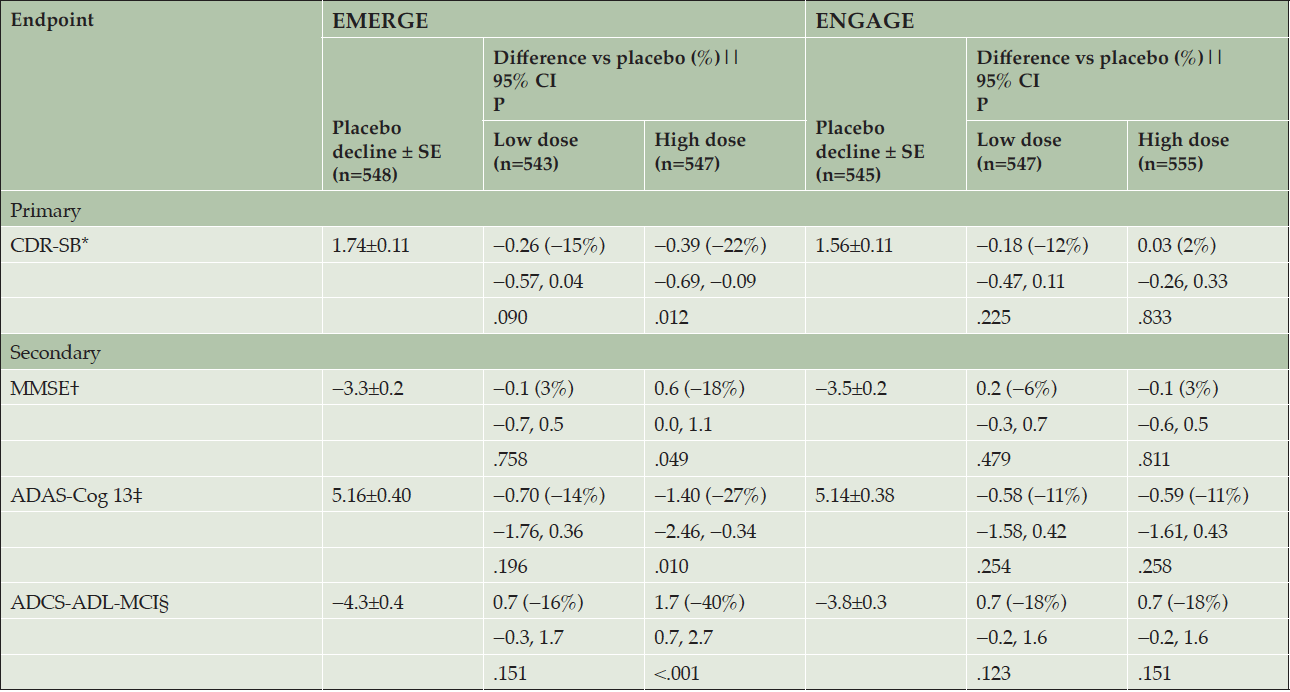
Table 2. Primary and secondary endpoints at week 78
Randomized and dosed population; A mixed model for repeated measure model was used to assess CDR-SB, MMSE, ADAS-Cog13, and ADCS-ADL-MCI scores, with fixed effects of treatment, categorical visit, treatment-by-visit interaction, baseline score, baseline score-by-visit interaction, baseline MMSE score (same as baseline score in the MMSE model), Alzheimer’s disease symptomatic medication use at baseline, region, and ApoE ε4 status (carrier and noncarrier). Data collected after March 20, 2019, were excluded; * CDR-SB scores range from 0 to 18, with higher scores indicating greater impairment; † MMSE scores range from 0 to 30, with lower scores indicating greater impairment; ‡ ADAS-Cog13 scores range from 0 to 85, with higher scores indicating greater impairment; § ADCS-ADL-MCI scores range from 0 to 53, with lower scores indicating greater impairment; || Difference vs placebo at week 78. Negative percentage means less progression in the treated arm; ADAS-Cog13, Alzheimer’s Disease Assessment Scale–cognitive subscale (13 items); ADCS-ADL-MCI, Alzheimer’s Disease Cooperative Study–Activities of Daily Living Inventory, mild cognitive impairment version; ApoE, apolipoprotein E; CDR-SB, Clinical Dementia Rating Scale–sum of boxes; MMSE, Mini-Mental State Examination.
Results from the aducanumab low-dose arm of ENGAGE were generally consistent across prespecified subgroups (Supplemental Data Fig. 3m-p). High-dose aducanumab subgroup results in ENGAGE were more variable (Supplemental Data Fig. 3e-h).
Amyloid PET substudies assessed n=488 and n=585 patients in EMERGE and ENGAGE, respectively. These substudies showed a dose- and time-dependent reduction in amyloid PET SUVR in both EMERGE and ENGAGE. At week 78, the difference in adjusted mean change from baseline between high-dose aducanumab and placebo was −0.278 (95% CI, −0.306 to −0.250; P<.0001) for EMERGE (Fig. 2a) and −0.232 (95% CI, −0.256 to −0.208; P<.0001) for ENGAGE (Fig. 2b). For the high-dose aducanumab arm, the reduction in adjusted mean change from baseline in amyloid PET SUVR in ENGAGE was 16.5% less than that in EMERGE at week 78.
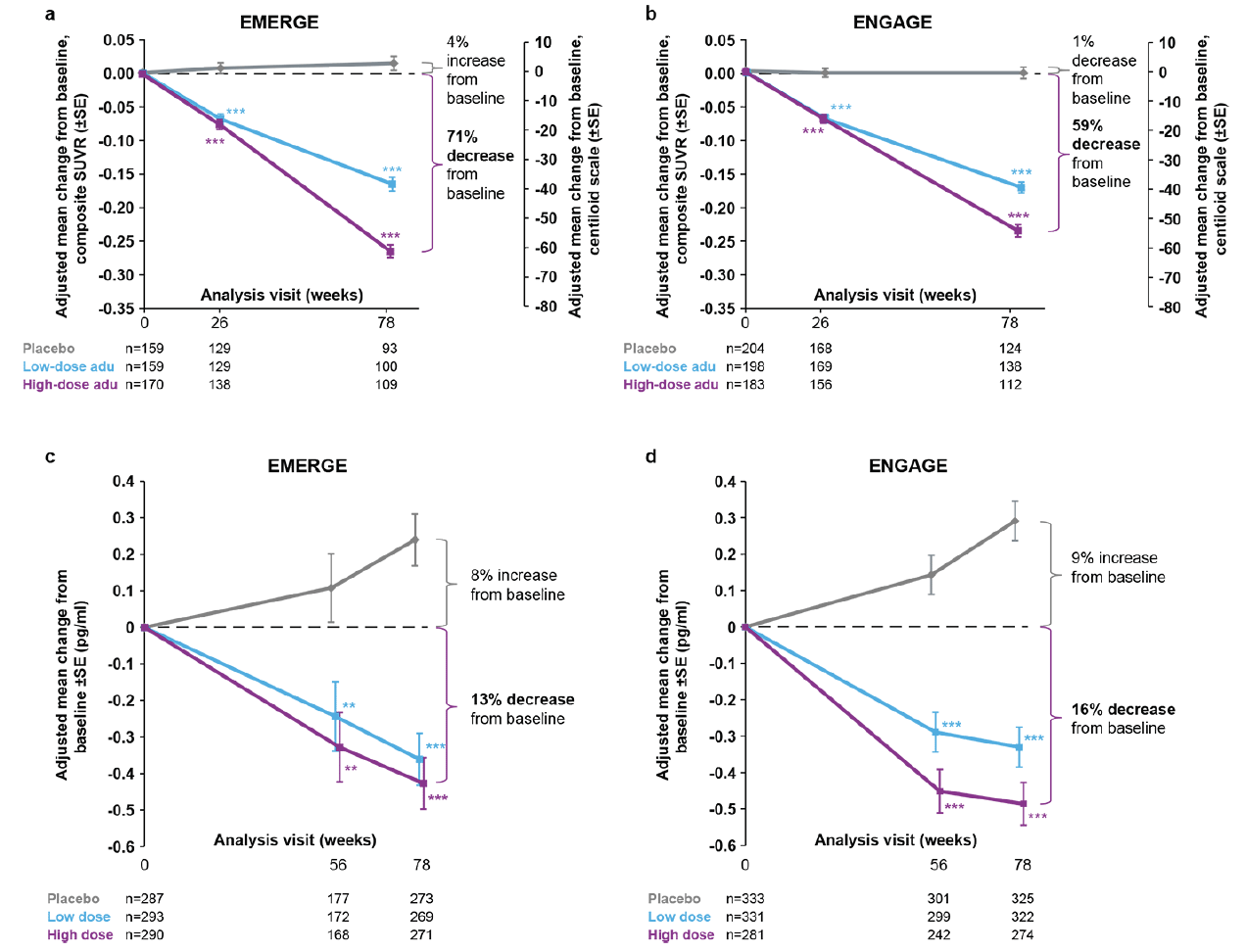
Figure 2. Longitudinal change from baseline in amyloid PET (composite SUVR and centiloid) and plasma p-tau181
Longitudinal change from baseline in amyloid PET composite SUVR (and centiloid) assessed by 18F-florbetapir in the amyloid PET substudies of EMERGE (a) and ENGAGE (b). In panels (a) and (b), percentages from baseline are based on the centiloid scale. The composite SUVR was computed from the frontal, parietal, lateral temporal, sensorimotor, anterior, and posterior cingulate cortices and normalized using the cerebellum as the reference region. Longitudinal change from baseline in plasma p-tau181 levels in the plasma p-tau181 analysis populations from EMERGE (c) and ENGAGE (d); **P<.01 *** P<.001. Error bars denote SE; adu, aducanumab; PET, positron emission tomography; SUVR, standardized uptake value ratio.
The adjusted mean changes from baseline in amyloid PET SUVR for low-dose aducanumab arms were similar between EMERGE and ENGAGE at week 78.
After 78 weeks, 48% of patients from EMERGE and 31% of patients from ENGAGE treated with high-dose aducanumab had a PET composite SUVR score of ≤1.10, a proposed threshold that distinguishes between Aβ-negative and -positive patients (Supplemental Data Table 4) (25).
Plasma p-tau was assayed in 870 and 945 patients in EMERGE and ENGAGE, respectively. An increase over time in plasma p-tau181 levels, was observed in the placebo groups of both EMERGE (Fig. 2c) and ENGAGE (Fig. 2d). In the treatment arms, reductions in plasma p-tau181 levels were observed over time. The difference in adjusted mean change from baseline between high-dose aducanumab and placebo was −0.667 (95% CI, −0.860 to −0.474; P<.0001) for EMERGE and −0.777 (95% CI, −0.931 to −0.623; P<.0001) for ENGAGE. More modest decreases were observed in the low-dose aducanumab groups. In both EMERGE and ENGAGE, reductions in plasma p-tau181 levels were positively correlated with reductions in amyloid PET SUVR at week 78 (Supplemental Data Fig. 4a).
Group-level correlation analyses based on data from aducanumab studies demonstrated a correlation in the hypothesized direction between treatment effects on Aβ PET and CDR-SB, indicating that a greater treatment effect on brain Aβ plaque levels was associated with a greater clinical benefit (Supplemental Data Fig. 4b).
Results of patient-level correlation analyses between change from baseline to week 78 amyloid PET composite SUVR and each of the four clinical measures (the primary endpoint and three secondary endpoints) in the combined low- and high-dose aducanumab–treated patients from each study are shown in Supplemental Data Fig. 4c. In EMERGE, modest correlations between amyloid PET SUVR and clinical endpoints were observed. In ENGAGE, in which a clinical treatment effect was not observed, correlations were not apparent.
The relationship between the aducanumab-induced treatment effect on plasma p-tau181 levels and clinical decline was also examined (Supplemental Data Fig. 4c). Correlations in the hypothesized direction were observed in the aducanumab-treated groups in both EMERGE and ENGAGE, indicating that a greater reduction in plasma p-tau181 levels was associated with less clinical decline.
In the EMERGE and ENGAGE CSF substudies (Supplemental Data Fig. 5a,b), a dose-dependent increase in CSF Aβ1-42 levels was observed along with a dose-dependent decrease in CSF p-tau and t-tau levels in EMERGE; in ENGAGE, CSF Aβ1-42 level was increased in the high-dose group, while a numerical decrease was observed in the high-dose group for CSF p-tau and t-tau levels. Although sample sizes were small, pooled results from EMERGE and ENGAGE demonstrated a reduction in tau PET signal in the medial temporal, temporal, and frontal lobes with high-dose aducanumab treatment (Supplemental Data Fig. 5c). A numerical trend of treatment effect favoring aducanumab was observed in the cingulate composite (low dose, −0.033; high dose, −0.015) and parietal composite (low dose, −0.046; high dose, −0.048); a numerical trend favoring placebo was observed in the occipital composite (low dose, 0.004; high dose, 0.018).
The effect of aducanumab on structural MRI, a measure of neurodegeneration, was also assessed (Supplemental Data Fig. 5d,e). A significant increase in the change from baseline to week 30 and week 78 in MRI lateral ventricle volume was observed in all aducanumab treatment groups (low- and high-dose groups in both EMERGE and ENGAGE) relative to placebo (P<.0001); no effects related to treatment were observed in measures for hippocampus and whole brain).
Safety
The incidence of adverse events was similar across dose groups in both studies (Table 3). Except for ARIA, the incidence and type of adverse events were consistent with those expected in an AD population (18). Sixteen deaths occurred across the two studies (n=5 in placebo; n=3 in low-dose aducanumab; n=8 in high-dose aducanumab), none of which were attributed by the investigator to study treatment.
Adverse events with an incidence >10% in any dose group were ARIA-E, headache, brain microhemorrhages (ARIA-H microhemorrhage), nasopharyngitis, fall, localized superficial siderosis (ARIA-H superficial siderosis), and dizziness (Table 3). The incidence of ARIA-E was higher in the high-dose groups compared with low-dose groups (35% vs 26%, respectively, in EMERGE and 36% vs 26% in ENGAGE) and higher in ApoE ε4 carrier levels compared with those of noncarriers (43% vs 18%, respectively, in the EMERGE high-dose group and 42% vs 23% in the ENGAGE high-dose group). In the combined high-dose groups, the incidence of ARIA-E was 65% in homozygous carriers and 35% in heterozygous carriers.
The majority of first ARIA-E events occurred early in treatment, during the first eight doses (EMERGE: 69.1%; ENGAGE: 77.4%). Of ARIA-E events , 98% resolved on study: EMERGE high-dose groups: 65% resolved within 12 weeks and 79% resolved within 16 weeks; ENGAGE high-dose groups: 72% resolved within 12 weeks and 85% resolved within 16 weeks. 10% of all patients had recurrent ARIA in the combined high-dose arms during the studies.
The incidences of brain microhemorrhages and localized superficial siderosis were higher in aducanumab-treated participants with ARIA-E compared with study participants without ARIA-E (brain microhemorrhages: 20% vs 9%, respectively, in the EMERGE high-dose group and 19% vs 6% in the ENGAGE high-dose group; localized superficial siderosis: 13% vs 2%, respectively, in the EMERGE high-dose group and 16% vs 1% in the ENGAGE high-dose group). The incidences of brain microhemorrhages and localized superficial siderosis in aducanumab-treated participants without ARIA-E were similar to the respective incidences in the placebo group (Table 3).
A minority of study participants with ARIA reported symptoms in the setting of an ARIA episode (EMERGE: 20% high dose, 21% low dose, 4% placebo; ENGAGE: 29% high dose, 16% low dose, 5% placebo). Symptoms reported during ARIA among participants in the high-dose groups of each study were typically transient and included headache (44% in EMERGE, 49% in ENGAGE), confusion (13% in EMERGE, 15% in ENGAGE), dizziness (11% in EMERGE, 12% in ENGAGE), and nausea (2% in EMERGE, 12% in ENGAGE).
Serious ARIA events were uncommon (EMERGE: 1.5% high dose, 0.9% low dose, and 0.2% placebo; ENGAGE: 1.4% high dose, 0.4% low dose, and 0.2% placebo); in the high-dose group, such events were observed in both ApoE ε4 carriers (EMERGE: 1.1%; ENGAGE 1.3%) and noncarriers (EMERGE: 2.2%; ENGAGE 1.4%). Serious events reported as symptoms of ARIA were confusional state, delirium, gait disturbance, generalized tonic-clonic seizure, memory impairment, seizure, and headache (Supplemental Data Table 5). Severe AEs that investigators reported as ARIA symptoms included headache, confusional state, seizure, and muscle weakness due to cerebral hemorrhage. The most common severe symptom among aducanumab-treated patients was headache (n=4); all other symptoms occurred in one patient each (Supplemental Data Table 6). There were no fatal events due to ARIA in either study.
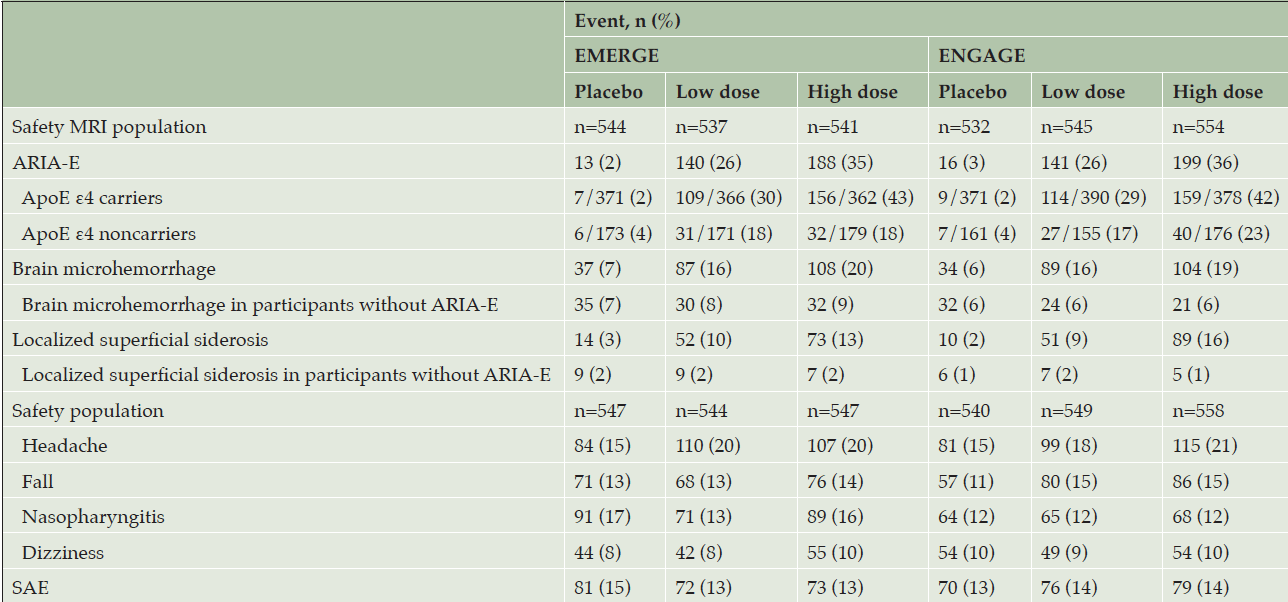
Table 3. Summary of adverse events
The safety MRI population denotes all randomized participants who received at least one dose of study treatment and had at least one postbaseline MRI assessment. The safety population denotes all randomized participants who received at least one dose of study treatment; ApoE, apolipoprotein E; ARIA-E, amyloid related imaging abnormalities-edema; MRI, magnetic resonance imaging, SAE, serious adverse event.
Discussion
The EMERGE and ENGAGE trials were terminated early due to the outcome of a futility analysis. Futility analyses are included in clinical studies to prevent participants from receiving ineffective treatments. These analyses can, however, have important limitations. In this case, two key assumptions were violated: 1) the assumption that the treatment effect in the two studies would be similar and 2) the assumption that the treatment effect would not change substantially over time. In fact, the treatment difference vs. placebo differed between studies, and both studies showed a larger magnitude of treatment effect in the final data compared with the futility interim data. The second assumption, constancy of effect, was further challenged by protocol amendments (see Methods) that changed target dose for approximately two-thirds of the high-dose aducanumab group partway through the studies. Unfortunately, at the time of futility, these assumptions were not assessed and should have been verified in hindsight. In general, conducting a futility analysis lowers the chance of detecting a positive result at final analysis.
Given early termination of the studies, it is reasonable to question the validity of the study results. However, no evidence has shown that the early termination of the studies affected the integrity or validity of the results or conclusions from either study. All data collected up until the futility announcement was collected under unchanged, protocol-specified, double-blind conditions, as the studies continued to be conducted per clinical study protocols. The final data were analyzed based on the prespecified analysis plan except for one change: to exclude data collected on or after the futility announcement on March 21, 2019. The rationale for this change was a conservative approach to minimize potential bias by excluding any data that might be impacted at the time of collection by the announcement of futility. Conclusions related to the primary and secondary efficacy outcomes were based on prespecified analyses with hierarchical testing and not on post hoc analyses. Furthermore, because no superiority analysis was conducted, there was no alpha spent because of the interim futility analysis. Early termination of the studies did, however, result in fewer data on which to perform the analyses than was initially planned. Several sensitivity and supplementary analyses were conducted to evaluate the impact of the missing data caused by early study termination (Supplemental Data Table 2). These analyses yielded similar results to the primary analysis, demonstrating the robustness of the study results.
The results from the prespecified primary and secondary clinical endpoints in EMERGE and ENGAGE were partially discordant. In ENGAGE, the primary and secondary endpoints were not met. In EMERGE, a statistically significant slowing of clinical decline was seen in the high-dose arm for the primary endpoint (CDR-SB) and three secondary endpoints (MMSE, ADAS-Cog13, and ADCS-ADL-MCI), demonstrating a consistent benefit of high-dose aducanumab over placebo. The findings from the low-dose groups in both studies were similar in magnitude and numerically favored aducanumab.
It is highly unlikely and unexpected to have two studies show contradictory clinical outcomes: one with negative, and one with statistically significant and internally consistent results. The EMERGE findings are unlikely to be false-positive; results are highly internally consistent across diverse clinical endpoints and subgroups. The aducanumab high-dose arm demonstrated a statistically significant effect on the primary and all three secondary endpoints, with all tests satisfying the prespecified multiple testing procedure and a nominally statistically significant effect on the tertiary clinical efficacy endpoint (NPI-10). Furthermore, results were robust to departures from assumptions regarding missing data and non-normality, and 79 of 80 subgroup comparisons showed a numerical advantage of aducanumab over placebo (Supplemental Data Fig. 3a-d). These findings are consistent with emerging clinical data seen in the phase 2 trials of two other anti-Aβ mAbs (10, 28).
ENGAGE is a negative study, with the primary endpoint not met. Consequently, per the statistical analysis plan, all testing of endpoints ceases there. However, to understand ENGAGE results, it is essential to look at data from all endpoints. In ENGAGE, across high- and low- dose arms, three of 10 clinical endpoints did not directionally favor an aducanumab treatment effect. The high-dose group in ENGAGE performed numerically worse than the low-dose group on the primary outcome (2%); this was also observed on the MMSE (3%). Across the EMERGE and ENGAGE studies and across low and high-dose groups, 16 of 20 largely independent clinical results directionally favor an aducanumab treatment effect. Thus, the clinical results of the two trials were partially discordant.
Aducanumab selectively targets aggregated forms of Aβ and has previously shown dose- and time-dependent reduction in Aβ plaques (8). In Aβ PET substudies, significant dose- and time-dependent reductions in amyloid PET SUVR were associated with aducanumab treatment in both EMERGE and ENGAGE. However, the magnitude of these changes differed in the high-dose arms: reductions in brain amyloid levels were 16.5% lower at week 78 in ENGAGE (−0.232) compared with EMERGE (−0.278).
Effects on downstream biomarkers specific to AD (tau PET, CSF p-tau, and plasma p-tau181) were also observed in both studies. Dose-related decreases in CSF p-tau levels were observed in both trials; the differences vs placebo were significant in EMERGE and numerical in ENGAGE. Reductions in the levels of plasma p-tau181, a newly established marker of soluble p-tau in the brain and AD progression (26), were observed over time in both studies. Additionally, pooled results from a small sample size of EMERGE and ENGAGE participants also demonstrated dose-dependent reductions of tau PET SUVR in the medial temporal, temporal, and frontal lobes.
Biomarker changes indicative of AD are ordered temporally over the course of disease, with recent data suggesting that an increase in Aβ plaques precedes an increase in the levels of soluble p-tau, which in turn may drive the accumulation of neurofibrillary tangles (NFTs) and subsequent cognitive decline (29). These findings from EMERGE and ENGAGE demonstrate that treatment with aducanumab, an anti-Aβ monoclonal antibody, directly affects both an upstream biomarker of AD (Aβ plaque) as well as an intermediate biomarker of AD (soluble p-tau). In both studies, reductions in amyloid PET SUVR were correlated with a reduction in plasma p-tau181 levels. Additionally, a reduction in the levels of each of these biomarkers was generally associated with less clinical decline across aducanumab studies (27). In group-level correlation between amyloid PET SUVR and clinical decline, the high-dose arm in ENGAGE was the only group that deviated from the overall trend. Together, these results support the hypothesis that Aβ accumulation triggers downstream tau pathology and subsequent clinical decline and that targeting aggregated Aβ in the brain via aducanumab treatment could result in clinical benefit. Emerging clinical trial data from several new anti-Aβ mAbs provide additional support for this mechanistic hypothesis (10, 27, 28, 30).
Rates of change in several structural measures, including whole brain and hippocampus, as well as ventricular enlargement, correlate with changes in cognitive performance, supporting their validity as markers of disease progression (31). In EMERGE and ENGAGE, progression of atrophy was seen by MRI; however, no effects related to treatment were observed in hippocampus or whole brain. A significant but very small increase, a difference of < 0.2% of the total intracranial space, in ventricular volume was observed in both low- and high-dose aducanumab treatment groups in both EMERGE and ENGAGE) relative to placebo (P<.0001). An increase in lateral ventricular volume has also been noted with other anti-amyloid therapies and is thought to be due to factors other than neurodegeneration (32).
Collectively, the biomarker results from EMERGE and ENGAGE demonstrate a consistent modification of underlying pathophysiology of disease with aducanumab treatment, whereas the clinical results were discordant for the high-dose arms of the two studies. It is not unexpected that effects on upstream biomarkers of disease pathophysiology may be detectable within a shorter treatment window as compared with downstream measures of disease, such as clinical symptomology.
The safety profiles of aducanumab in EMERGE and ENGAGE were consistent with each other and consistent with those of previous aducanumab studies (8, 11). The most common adverse event associated with aducanumab was ARIA-E (12), an imaging abnormality detected via brain MRI in both studies. Serious and severe symptoms did occur in the setting of ARIA-E, including seizures, which on rare occasions required hospitalization. ARIA-E is an important adverse event of amyloid-lowering therapies that is important to monitor and manage during treatment (33).
While EMERGE and ENGAGE were identical in design, the implementation of the studies was not. Many elements, such as demographic and disease characteristics, as well as frequency, severity, and management of ARIA, were similar between the studies and did not appear to account for the partially discordant clinical results between the two studies. Although ARIA has the potential to functionally unblind patients and caregivers, no systematic bias caused by ARIA could be detected in either study. However, as described in the Methods, two protocol amendments (PV3 and PV4) allowed more participants in the high-dose groups to achieve the target dose of 10 mg/kg (29% of patients in EMERGE and 22% of patients in ENGAGE received the full possible 14 doses of 10 mg/kg aducanumab). Due to differences in the rates of enrollment, ENGAGE had enrolled approximately 200 more participants than EMERGE at the time of the PV4 amendment (Supplemental Data Fig. 1a). Given that previous clinical and nonclinical studies of aducanumab showed a clear dose-exposure response (8, 11), it is of high interest to determine the extent to which differential dosing contributed to the discordant findings in the high-dose arms of EMERGE and ENGAGE. This, as well as further analyses of other potential key factors that may have contributed to the discordant clinical findings in the high-dose groups will be the focus of a forthcoming manuscript.
Among the limitations of these studies are the invalid assumptions underpinning the futility decision, which resulted in early termination based on an inaccurate prediction of the final outcomes. An additional limitation of these studies is that some of the biomarker results should be interpreted with caution; the CSF and tau PET biomarker substudies had relatively small sample sizes from a nonrandom subset of trial participants (i.e., those who chose to opt in to each substudy). Overall, the populations in these studies lack diversity, including racial/ethnic diversity, patients with co-morbid conditions, and those on some concomitant medications. The sponsor, Biogen, recognizes that the Black/African American and Hispanic populations in the EMERGE and ENGAGE trials are not representative of the community. This limits the generalizability of the data and additional data generation is required. An enrollment target of 18% Black and Hispanic Americans has been set for planned trials. Collection of additional data within controlled clinical studies and in the real-world setting using registry-based studies is on-going (https://clinicaltrials.gov/ct2/show/NCT0509713).
In summary, ENGAGE did not meet its primary or secondary endpoints, and the EMERGE high-dose aducanumab group met all primary and secondary endpoints. EMERGE is the first phase 3 trial to demonstrate an association between reduction of biomarkers of AD pathology and a statistically significant slowing of clinical decline, supporting the possibility that removal of Aβ from the brain (together with modification of downstream biomarkers of disease) may be associated with a clinical benefit in patients with early AD. The safety profile of aducanumab in EMERGE and ENGAGE was consistent with that of previous aducanumab studies, and a detailed investigation of ARIA has been published (12). Clinical efficacy of aducanumab will be further evaluated in a forthcoming clinical trial.
Funding: This study was sponsored by Biogen. Biogen played a role in the design and conduct of the study as well as the collection, analysis, and interpretation of data and Biogen authors played a role in the preparation of this manuscript. Medical writing support, under direction of the authors, was provided by Meditech Media and was funded by Biogen.
Acknowledgments: The authors thank the participants and their family members in the aducanumab studies, as well as the investigators and staff conducting these studies. We thank John O’Gorman for his contribution to the study design and protocol. We thank Kathleen Grobben, Tucker Ward, Ramon Iovin, and Eric Ponton for editorial support and coordination of the authors in the development of this manuscript.
Author contributions: All authors made substantial contributions to the acquisition, analysis, or interpretation of the data; critically revised the manuscript for intellectual content; approved the final version to be published; and are accountable for all aspects of the work.
Competing interest declaration: These studies were funded by Biogen. SBH, YT, KKM, TC, SW, GD, KH, IN, and YZ are employees and shareholders of Biogen. SC (Dr Chalkias), AS, LN, RR, CvH, CM and LS were employees of Biogen at the time of this work. CM is a current employee of Cortexyme. LS and CvH are current employees of Takeda Pharmaceutical Co. SC is a current employee at Moderna. RR is a current employee at Vigil Neuro. PSA was chair of the Steering Committee and has consulted for Biogen, has received research support from Eli Lilly, Janssen, Eisai, the Alzheimer’s Association, the National Institutes of Health, and the Foundation for the National Institutes of Health; he has consulted for Merck, Roche and ImmunoBrain Checkpoint. PSA is co-editor in chief of JPAD with no personal compensation; he did not have a role in the editorial process/review for this manuscript. FB was supported by NIHR Biomedical Research Centre at UCLH. He receives personal fees for consultancy from Bayer, Biogen, Roche, IXICO Ltd, Novartis, and Combinostics. SC (Dr Cohen) was an ENGAGE trial site investigator and an Aducanumab Steering Committee member. She is a consultant (no personal fees received) to Biogen, Cassava Sciences, Cogstate, INmune Bio, ProMIS Neuroscience, and RetiSpec and receives research support (paid to institution) from AgeneBio, Anavex, Biogen, Cassava, Eisai, Genentech, Green Valley, Eli Lilly, Janssen, RetiSpec, Roche, and Vielight. OH received research support (for the institution) from AVID Radiopharmaceuticals, Biogen, Eli Lilly, Eisai, GE Healthcare, Pfizer, and Roche. In the past 2 years, he has received consultancy/speaker fees from AC Immune, Alzpath, Biogen, Cerveau, and Roche. TI is a consultant to Eisai and Roche. CJM was an ENGAGE trial site investigator and an Aducanumab Steering Committee member. She is supported by NIHR Biomedical Research Centre at UCLH and has acted as a consultant to Biogen, Roche, and IONIS. CvD was an EMERGE trial site investigator. He is a consultant for Roche, Eisai, and Ono and receives research support from Biogen, Eisai, Roche, Genentech, Eli Lilly, Janssen, Merck, Novartis, and Biohaven. BV is an investigator in clinical trials sponsored by Biogen, Lilly, Roche, Eisai Pharmaceuticals, and the Toulouse University Hospital (Inspire Geroscience Program). He has served as SAB member for Biogen, Alzheon, Green Valey, Norvo Nordisk, Longeveron, but received no personal compensation. He has served as consultant and/or SAB member for Roche, Lilly, Eisai, TauX with personal compensation. His family members have equity ownership interest in Serdi. He is member of the Editorial Board of JPAD with no personal compensation. He did not have a role in the editorial or peer review for this manuscript at JPAD.
Ethical standard: The study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization and Good Clinical Practice guidelines and was approved by ethics committees or institutional review boards at each site. All patients provided written informed consent. An independent data monitoring committee was responsible for monitoring the conduct of the trials, assessing routine safety, and reviewing futility analysis results.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. Jack CR Jr, Bennett DA, Blennow K, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018;14:535-562. DOI: https://doi.org/10.1016/j.jalz.2018.02.018
2. Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science 1992;256:184-185. DOI: https://doi.org/10.1126/science.1566067
3. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 2016;8:595-608. DOI: https://doi.org/10.15252/emmm.201606210
4. Cummings J, Goldman DP, Simmons-Stern NR, Ponton E. The costs of developing treatments for Alzheimer’s disease: A retrospective exploration. Alzheimers Dement. 2021;1-9. DOI: https://doi.org/10.1002/alz.12450
5. Liu E, Schmidt ME, Margolin R, et al. Amyloid-β 11C-PiB-PET imaging results from 2 randomized bapineuzumab phase 3 AD trials. Neurology 2015;85:692-700. DOI: https://doi.org/10.1212/WNL.0000000000001877
6. Salloway S, Honigberg LA, Cho W, et al. Amyloid positron emission tomography and cerebrospinal fluid results from a crenezumab anti-amyloid-beta antibody double-blind, placebo-controlled, randomized phase II study in mild-to-moderate Alzheimer’s disease (BLAZE). Alzheimers Res. Ther. 2018;10:96. DOI: https://doi.org/10.1186/s13195-018-0424-5
7. Siemers ER, Sundell KL, Carlson C, et al. Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement. 2016;12:110-120. DOI: https://doi.org/10.1016/j.jalz.2015.06.1893
8. Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016;537:50-56. DOI: https://doi.org/10.1038/nature19323
9. Swanson CJ, Zhang Y, Dhadda S, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res. Ther. 2021;13:80. DOI: https://doi.org/10.1186/s13195-021-00813-8
10. Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer’s disease. N Engl J Med. 2021;384:1691-1704. DOI: https://doi.org/10.1056/NEJMoa2100708
11. ADUHELM [prescribing information]. Cambridge, MA: Biogen, Inc. 2021.
12 Salloway S, Chalkias S, Barkhof F, et al. Amyloid-related imaging abnormalities in 2 Phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol. 2022;79:13-21. DOI: https://doi.org/10.1001/jamaneurol.2021.4161
13. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270-279. DOI: https://doi.org/10.1016/j.jalz.2011.03.008
14. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263-269. DOI: https://doi.org/10.1016/j.jalz.2011.03.005
15 U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation. Early Alzheimer’s disease: developing drugs for treatment. In: Guidelines for Industry. 2018. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM596728.pdf. Accessed December 29, 2021
16. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975;12:189-198. DOI: https://doi.org/10.1016/0022-3956(75)90026-6
17. Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412-2414. DOI: https://doi.org/10.1212/wnl.43.11.2412-a
18. Sperling RA, Jack CR Jr, Black SE, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7:367-385. DOI: https://doi.org/10.1016/j.jalz.2011.05.2351
19. Mohs RC, Knopman D, Petersen RC, et al. Development of cognitive instruments for use in clinical trials of antidementia drugs: additions to the Alzheimer’s Disease Assessment Scale that broaden its scope. The Alzheimer’s Disease Cooperative Study. Alzheimer Dis. Assoc. Disord. 1997;11 Suppl 2:S13-21. DOI: https://doi.org/10.1097/00002093-199700112-00003
20. Galasko D, Bennett D, Sano M, et al. An inventory to assess activities of daily living for clinical trials in Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. Alzheimer Dis Assoc Disord 1997;11 Suppl 2:S33-39. DOI: https://doi.org/10.1097/00002093-199700112-00005
21. Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology 1994;44:2308-2314. DOI: https://doi.org/10.1212/wnl.44.12.2308
22. Ostrowitzki S, Deptula D, Thurfjell L, et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch. Neurol. 2012;69:198-207. DOI: https://doi.org/10.1001/archneurol.2011.1538
23. Klunk WE, Koeppe RA, Price JC, et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement 2015;11:1-15.e154. DOI: https://doi.org/10.1016/j.jalz.2014.07.003
24. Deng Q, Zhang YY, Roy D, Chen MH. Superiority of combining two independent trials in interim futility analysis. Stat Methods Med Res 2020;29:522-540. DOI: https://doi.org/10.1177/0962280219840383
25. Joshi AD, Pontecorvo MJ, Lu M, Skovronsky DM, Mintun MA, Devous MD Sr. A semiautomated method for quantification of F 18 Florbetapir PET images. J Nucl Med. 2015;56:1736-41. DOI: https://doi.org/10.2967/jnumed.114.153494
26. Janelidze S, Mattsson N, Palmqvist S, et al. Plasma p-tau181 in Alzheimer’s disease: Relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med. 2020;26:379-86. DOI: https://doi.org/10.1038/s41591-020-0755-1
27. Center for Drug Evaluation and Research. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/761178Orig1s000ClinPharm_Redacted.pdf. Accessed December 29, 2021.
28. Swanson CJ, et al. Oral presentation at: 11th Annual Clinical Trials on Alzheimer’s Disease; October 24-27, 2018; Barcelona, Spain.
29. Hansson O. Biomarkers for neurodegenerative diseases. Nature Medicine. 2021;27:954-963. DOI: https://doi.org/10.1038/s41591-021-01382-x
30. Salloway S, Farlow M, McDade E, et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat Med. 2021;27:1187-1196. DOI: https://doi.org/10.1038/s41591-021-01369-8
31. Frisoni GB, Fox NC, Jack CR Jr, Scheltens P, Thompson PM. The clinical use of structural MRI in Alzheimer disease. Nat Rev Neurol. 2010;6:67-77. DOI: https://doi.org/10.1038/nrneurol.2009.215
32. Novak G, Fox N, Clegg S, et al. Changes in brain volume with bapineuzumab in mild to moderate Alzheimer’s disease. J Alzheimers Dis. 2016;49:1123-1134. DOI: https://doi.org/10.3233/JAD-150448
33. Cummings J, Aisen P, Apostolova LG, Atri A, Salloway S, Weiner M. Aducanumab: Appropriate use recommendations. J Prev Alzheimers Dis 2021;8:398-410. DOI: https://doi.org/10.14283/jpad.2021.41