
W. Luca, K. Foster, K. McClure, M.K. Ahlijanian, M. Jefson
Pinteon Therapeutics, Inc., 1188 Centre Street, Newton Centre, MA 02459, USA
Corresponding Author: Wendy Luca, 1188 Centre Street, Newton Centre, MA 02459, USA, +1 860-319-9938, wluca@pinteon.com
J Prev Alz Dis 2024;2(11):366-374
Published online January 24, 2024, http://dx.doi.org/10.14283/jpad.2024.25
Abstract
BACKGROUND: PNT001 is a humanized full-length IgG4 S228P monoclonal antibody that binds the cis conformation of the phosphorylated Thr231-Pro232 motif in human full-length (2N4R) tau (cis-pT231 tau) with high selectivity and affinity. It binds selectively to cis-pT231 tau in human tauopathy brain sections, inhibits aggregation of tau, and has shown efficacy in preclinical models of tauopathy. Good Laboratory Practice six-month toxicology studies in cynomolgous monkeys have shown no test article-related findings.
OBJECTIVES: To evaluate the safety, tolerability, pharmacokinetics, and immunogenicity of single escalating intravenous doses of PNT001 in healthy volunteers.
DESIGN: Phase 1, randomized, double-blind, and placebo-controlled 16-week study.
SETTING: Subjects were recruited across three clinical research sites in the United States.
PARTICIPANTS: Fifty healthy volunteer subjects enrolled, with 49 receiving the double-blind study drug.
INTERVENTION: Six cohorts were administered single escalating doses of PNT001 (33, 100, 300, 900, 2,700, and 4,000 mg). The subjects were randomized 6:2 (PNT001:placebo).
MEASUREMENTS: Safety was evaluated by the occurrence of adverse events, electrocardiography, physical examinations, neurological examinations, vital signs, and suicidality. Pharmacokinetics and biomarkers were assessed via serum and cerebrospinal fluid sample analyses.
RESULTS: Dose continuation after review of sentinel group data and dose escalation after completion of full cohort data were determined by an external, independent safety board. There were no study pauses or safety concerns identified by the safety board. A total of 49 subjects received the study drugs, with 36 receiving PNT001 and 13 receiving placebo. There were three related non-serious adverse events, each Grade 1, which occurred at the lowest doses and resolved without sequelae. No maximum tolerated dose was identified, and no premature discontinuations, dose reductions, or interruptions due to treatment-related adverse events occurred. One unrelated serious adverse event occurred in a placebo subject with an undisclosed medical condition. No other safety findings were identified. Doses of 900–4,000 mg produced concentrations in the cerebrospinal fluid exceeding the binding affinity constant of PNT001 for cis-pT231 tau (45 ng/mL), indicating that concentrations sufficient for target engagement can be obtained in the cerebrospinal fluid within the tested dose range. The serum pharmacokinetic profile was as expected for a monoclonal antibody. The terminal half-lives ranged from 23.8–33.8 days, and the cerebrospinal fluid exposures were approximately 0.1% of the plasma concentration and dose-proportional. Of the 36 subjects receiving PNT001, one post-baseline positive anti-drug antibody result was observed at Day 112 in a subject who received PNT001 (300 mg).
CONCLUSIONS: Single doses of PNT001 were safe and well-tolerated at all dose levels studied, including those doses expected to produce therapeutic benefit. These results support multiple ascending dose trials in patients with neurodegenerative tauopathies for this novel mid-domain tau antibody.
Key words: PNT001, cis pT231, mid-domain tau antibody.
Introduction
Extensive preclinical data indicate that the cis-conformer of tau that is phosphorylated at amino acid residue threonine-231 (cis-pT231 tau) has an important pathologic role in several tauopathies, including progressive supranuclear palsy (PSP), corticobasilar degeneration (CBD), Alzheimer’s disease (AD), and traumatic brain injury (TBI), and is therefore a plausible target for the treatment of both acute and chronic tauopathies (1-6).
PNT001 is a humanized full-length IgG4 S228P monoclonal antibody that binds the cis isomer of the phosphorylated mid-domain Thr231-Pro232 motif in human full-length (2N4R) tau (cis-pT231 tau) with >1,000-fold selectivity for the cis isomer of pT231 compared to the trans isomer and high affinity for cis-pTau231 (KD = 0.3 nM, ca. 45 ng/mL) (7). PNT001 localizes to neurofibrillary tangle-like structures in the brains of patients with PSP, AD, and chronic traumatic encephalopathy (CTE), with very limited reactivity in the brain tissue derived from age-matched controls (7).
A murine version of PNT001 containing identical complementarity in the determining regions to PNT001 (mPNT001) was tested in the Tg4510 mouse that overexpresses a pathologic, human mutant form of tau. mPNT001 significantly improved several endpoints in this study, including spatial memory and long-term potentiation, and reduced markers of neuroinflammation, insoluble tau, and serum neurofilament light chain concentrations (7). In a pivotal, preclinical, six-month Good Laboratory Practice safety study, animals received 26 doses of 10, 30, or 100 mg/kg via weekly intravenous (i.v.) infusion, and serum and cerebrospinal fluid (CSF) were sampled throughout the dosing period and during the eight-week recovery period. No test article-related clinical observations were noted at any dose level, and the no observed adverse effect level (NOAEL) was reported as the highest dose tested, 100 mg/kg (7). These results support further clinical development of PNT001 in humans.
Methods
Objectives
The primary objective of this study was to evaluate the safety and tolerability of single-escalating i.v. doses of PNT001 in healthy volunteers. The secondary objective was to evaluate the single-dose pharmacokinetic (PK) profile of PNT001 after a single i.v. dose of PNT001.
Study Design and Treatment
The PNT001 study (https://clinicaltrials.gov/ct2/show/NCT04096287?term=PNT001&draw=2&rank=2) was conducted between September 17, 2019 and February 15, 2021 at three Phase 1 centers. This study was conducted in accordance with the protocol, applicable regulatory requirements, International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Harmonised Tripartite Guideline for Good Clinical Practice, the Declaration of Helsinki as well as Title 21 of the US Code of Federal Regulations, Parts 50, 56, and 312. The investigator or investigator designee obtained written informed consent from each subject (or the subject’s legally acceptable representative) before performing any trial-specific screening or baseline period evaluations.
PNT001-001 was a multicenter, double-blind, single-ascending-dose trial that evaluated safety and tolerability in 49 healthy volunteers across six dose cohorts with the following ascending dose levels of PNT001 per cohort: 33 mg, 100 mg, 300 mg, 900 mg, 2,700 mg, and 4,000 mg. Each cohort included eight subjects (six active, two placebo) who were administered a single dose of PNT001 or placebo.
Dose Selection
The initial dose of 33 mg was established according to Food and Drug Administration (FDA) guidance (8). The single dose of 33 mg (0.47 mg/kg) provided a large safety margin compared with the human-equivalent dose of 33 mg/kg that was determined from the 100 mg/kg NOAEL dose in nonhuman primate toxicology studies. Subsequent dose levels were increased in three-fold increments with planned doses of 100, 300, 900, and 2,700 mg. An additional dose cohort of 4,000 mg was studied.
The clinical data were reviewed by an independent Data Safety Monitoring Board (DSMB). Within each cohort, the DSMB reviewed the data from a sentinel group of two subjects (one PNT001, one placebo) before the remainder of the cohort was enrolled. Enrollment of the remaining six subjects (randomized as five PNT001, one placebo) occurred after recommendation by the DSMB. Following review of the seven days of sentinel group safety data and the data on the 14 days of exposure to PNT001 or placebo of the entire cohort, the DSMB provided guidance for enrollment of the complete cohort and movement to the next dose cohort level. The study incorporated clearly defined stopping rules to minimize subject risk. The dose-limiting toxicity for PNT001 was defined in the protocol as more than two Grade-2 adverse events (AEs) or one Grade-3 AE that was a change from baseline and determined to be related to PNT001.
Study Drug Administration
PNT001 was diluted to the appropriate volume and concentration using 5% dextrose. The placebo consisted of an identical volume of 5% dextrose. The unblinded pharmacist was responsible for preparing the study drug and providing it to the study team member in a blinded fashion for administration to the subject.
Procedures
The study medication was administered as a single i.v. infusion over 30 minutes (60 minutes for doses greater than 2,700 mg), which was followed by the collection of safety, tolerability, and PK data over 16 weeks. After the initial screening and laboratory assessment performed on Day -1, eligible subjects were admitted to the research unit on Day 1 (or Day -1 at the discretion of the Investigator), where they were domiciled for three nights (four nights if admitted on Day -1) with standardized meals provided during their inpatient stay. On Day 1, subjects were randomized to receive either active drug or placebo. Subjects were discharged on Day 4 and returned for an outpatient study visit on Day 5 (via phone or clinic visit) and study site visits on Days 7, 14, 28, 42, 56, 70, 84, 98, and 112.
The safety assessments included assessments of AEs, electrocardiograms, physical examinations, neurological examinations, vital signs, immunogenicity, and suicidality and were performed throughout the 16-week study.
PK Assessments
The PK assessments included serum PK analyses of blood samples collected before dosing, at the end of infusion (0 hour), and 0.5, 1, 2, 4, 8, 12, 24, 36, and 48 hours after the end of infusion. Additional samples were collected on Days 7, 14, 28, 42, 56, 70, 84, 98, and 112. A saline or heparin lock was used to facilitate serial sampling on Days 1–3.
CSF samples for PK parameter assessments were collected on Days 3 and 28. If a subject terminated trial participation before Day 28, the CSF sample was collected at the End-of-Treatment visit for subjects who consented. Blood samples were processed to serum prior to analysis. The serum and CSF concentrations of PNT001 were measured using validated ligand binding assays (electrochemiluminescence for CSF, enzyme-linked immunosorbent assay for serum).
PK methods
The PK parameters were determined from the serum concentrations of PNT001 using non-compartmental analysis performed using Phoenix WinNonlin Version 8.1 (Certara USA, Inc., Princeton, NJ). PK analyses were conducted using actual post-dose times relative to the start time of dose administration recorded in the raw data. Statistical analyses were conducted to determine the dose proportionality of PNT001 for Area Under the Curve (AUC)0-t, AUC0-∞, and Cmax. The PK parameters were analyzed using the following power model:
parameter = intercept × dose^slope + random error
Using the natural log (ln) transformation, a power model can be expressed as a linear regression equation:
ln(parameter) = intercept + slope × ln(dose) + random error
For dose proportionality, the slope of the regression line would be equal to 1; for dose independence, it would be equal to 0.
For each PK parameter, a pooled estimate (across all doses) of the slope and the corresponding 95% confidence interval (CI) were calculated separately. Figures (on the logarithmic scale) containing the individual values, power model line (95% CI), and dose proportionality line (defined as the power model line with slope of 1) were created for each PK parameter; figures (on the semi logarithmic scale) containing individual values and geometric means were created for each corresponding PK parameter normalized by the dose administered.
A PK parameter was considered dose proportional for the dose range studied if the assumption of linearity was ruled acceptable and the 95% CI for the slope spanned 1. If the assumption of linearity was ruled unacceptable for any PK parameter, its corresponding PK parameter normalized by dose administered was ln-transformed and analyzed using an analysis of variance (ANOVA) model. The model included dose as a factor. For each PK parameter, the geometric least squares mean (GLSM) for each dose and the P values for the overall and pairwise dose comparisons were calculated separately. Residual plots were produced to assess the adequacy of the fitted model(s).
Entrance criteria
Eligible subjects included men and women who were 21 to 65 years old at the time of screening. Female subjects had documented proof that they were not of childbearing potential and were not breastfeeding. Male subjects agreed to use effective means of contraception. Subjects with medical or psychiatric conditions that could pose a safety risk or interfere with the study were excluded.
Statistical Methods
No formal statistical safety or efficacy analyses were planned for this study. The sample size was not based on statistical power considerations; however, the administration of PNT001 to six subjects per group has approximately 80% probability of observing one or more subjects with a serious AE (SAE) if the true incidence of an SAE is 24%. The summary tables of the data show the following: the number of subjects with non-missing data (n); the mean, standard deviation (SD), median, minimum, and maximum of the continuous data; and the counts and percentages of the categorical variables. The analysis sets and populations were defined as follows:
1. Full analysis set: all randomized subjects who received at least one dose of study drug.
2. Dose-limiting toxicity-evaluable analysis set: all enrolled subjects who received PNT001, had no major protocol violations, and completed the Day-14 visit or had a related Grade-3 or Grade-4 AE that was a change from baseline before or on Day 14.
3. Safety analysis set: all subjects who received the study drug and had at least one post-baseline safety assessment.
The serum PK population included all subjects who received the study medication and had sufficient serum concentration data for analysis. Based on the PNT001 concentration-time data, the following PK parameters were estimated using standard noncompartmental methods: AUC0-t, which was calculated by using the linear-log trapezoidal rule (the linear trapezoidal rule is applied up to maximum concentration and then the log trapezoidal rule is applied for the remainder of the curve, where t corresponds to the last measurable time point); AUC0-∞, which is equal to AUC0-t + Ct/λz, where Ct is the last measurable PNT001 concentration and λz is the terminal elimination rate constant calculated by using a log-linear regression of the terminal elimination phase of the serum concentration versus the time curve; Cmax of PNT001, which was determined from the observed PNT001 concentration-time data; time to maximum serum concentration (Tmax) of PNT001, which was calculated from the observed PNT001 concentration-time data, and the terminal half-life (t1/2), which was calculated as ln(2)/λz.
Results
Enrollment and Disposition
The subjects participated in the trial at three US sites from September 23, 2019, through February 15, 2021. A total of 50 subjects enrolled in the trial; one subject did not receive the study drug and was not included in the analyses. One subject received the study drug and was replaced. Thus, 49 subjects received the study drug. Of the 49, 36 received a single dose of PNT001, and 13 subjects received placebo. Of the 48 subjects that were not replaced, 41 (85.4%) completed the study. For the full and safety analysis populations, eight subjects (100%) were enrolled into each of the six dose cohorts (Cohort 1 [33 mg], Cohort 2 [100 mg], Cohort 3 [300 mg], Cohort 4 [900 mg], Cohort 5 [2,700 mg], and Cohort 6 [4,000 mg]). Nine subjects did not complete the trial due to loss to follow-up, physician decision, withdrawal by subject, or other reason. A summary of the subject enrollment and disposition information is provided in Table 1, Summary of Subject Enrollment and Disposition (All Randomized Subjects), and Figure 3, Enrollment and Disposition Diagram.
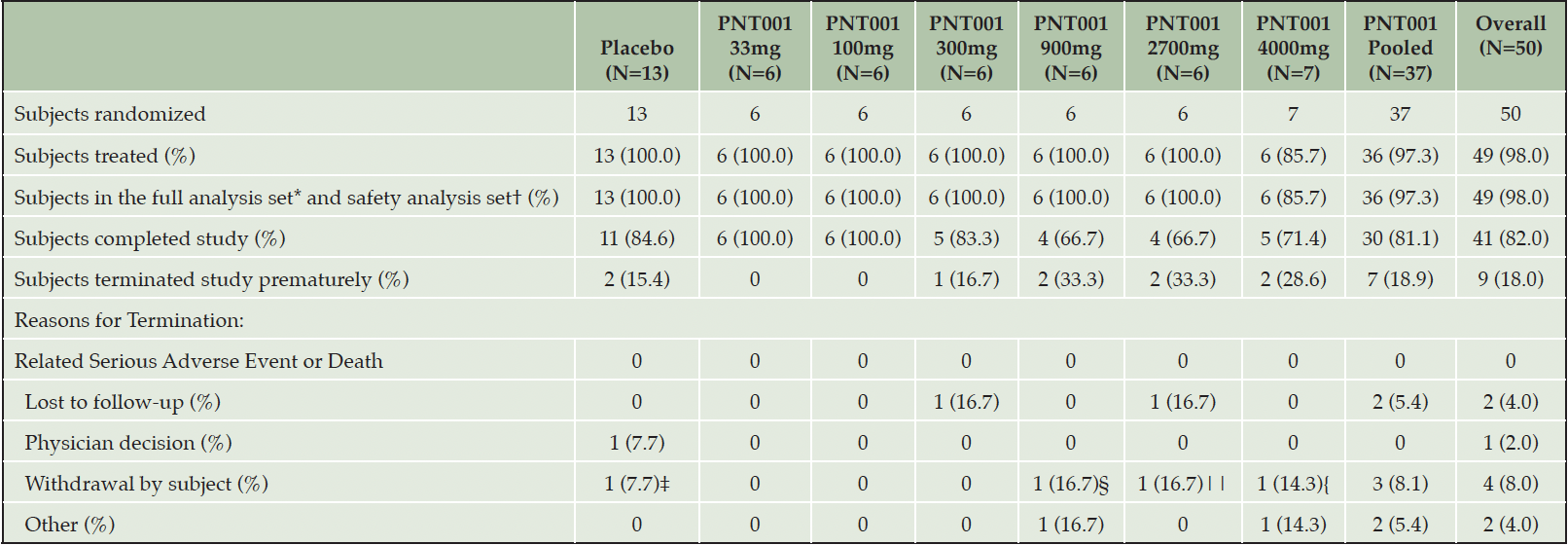
Table 1. Summary of Subject Enrollment and Disposition (All Randomized Subjects)
*The full analysis set (FAS) includes all randomized subjects who received at least one dose of study drug; †The safety population includes all subjects who received study drug and had at least one post-baseline safety assessment; ‡Subject 2022 did not wish to continue in the study. Mild adverse event of headache was reported. It is noted this discontinuation occurred March 12, 2020, at the beginning of the COVID-19 epidemic; §Subject 2030 left the unit on Day 2 stating they no longer wanted to participate. No adverse events were reported. It is noted the subject left the unit on April 3, 2020, at the beginning of the COVID-19 epidemic; ||Subject 2041 withdrew from the study. No specific reason is documented. No adverse events were reported. It is noted this discontinuation occurred July 28, 2020, in the early stages of the COVID-19 epidemic; {Subject 1047 withdrew from the study shortly after dosing after receiving a phone call notifying them of a family emergency.
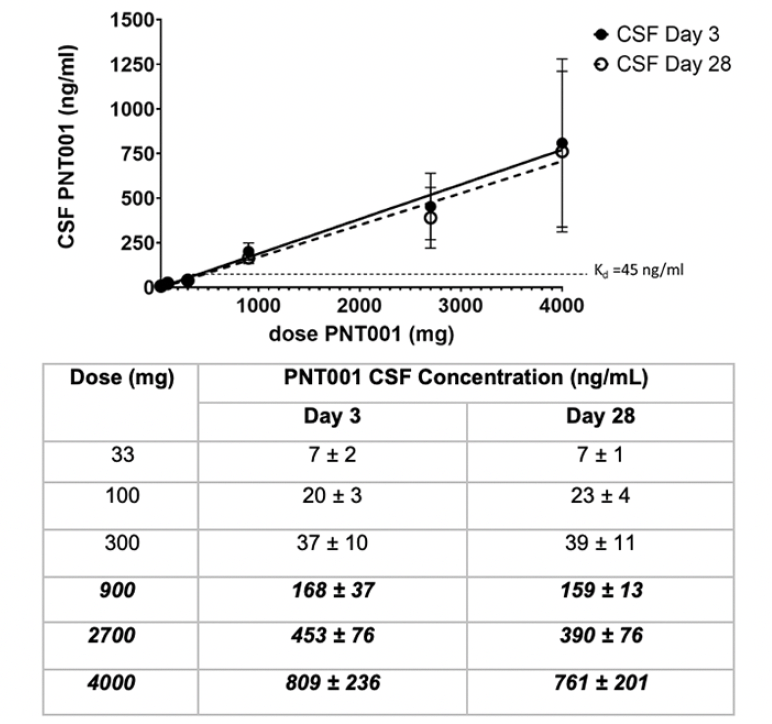
Figure 1. Arithmetic Mean (±Standard Deviation) Pharmacokinetic Concentration-time Profiles
Abbreviations: Tmax, time to maximum serum concentration; T1/2, terminal half-life, CL, clearance; Vd, volume of distribution.
Demographics and Baseline Characteristics
The overall mean (±SD) age of the subjects enrolled in the study was 47.6 (±11.9) years, and 73.5% were male. The racial distribution was 65.3% White, 30.6% Black, and 4.1% Asian. The overall mean height was 173.4 (±10.4) cm, the mean body weight was 80.7 (±13.0) kg, and the mean body mass index (BMI) was 26.7 (±2.5) kg/m2.
Treatment-Emergent Adverse Events (TEAEs)
Of the 49 subjects enrolled in the study who received the study drug, 22 (44.9%) subjects experienced at least one TEAE for a total of 35 events. Fourteen of the 36 (38.9%) subjects that received PNT001 and eight of the 13 (61.5%) subjects that received placebo reported at least one TEAE for a total of 22 and 13 events, respectively. Three (8.6%) TEAEs were determined to be related to PNT001. One (2.0%) subject experienced one unrelated SAE, and no subjects experienced a TEAE leading to early study termination or death.
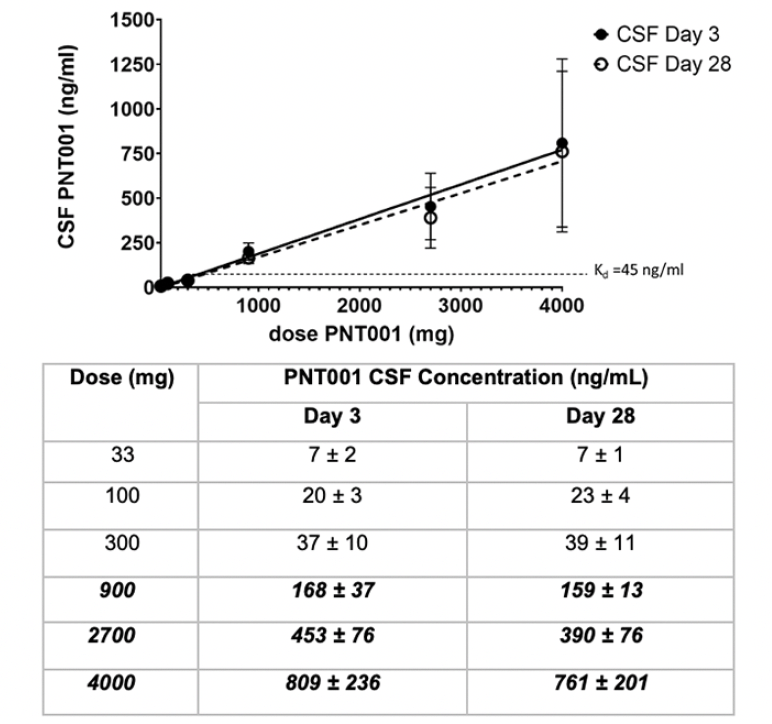
Figure 2. Cerebrospinal fluid (CSF) Concentrations ± Standard Deviation of PNT001 After Single Dose Administration by Dose Cohort
The bold italicized text indicates doses that resulted in CSF concentrations with dissociation constant (Kd) values over 45 ng/mL.
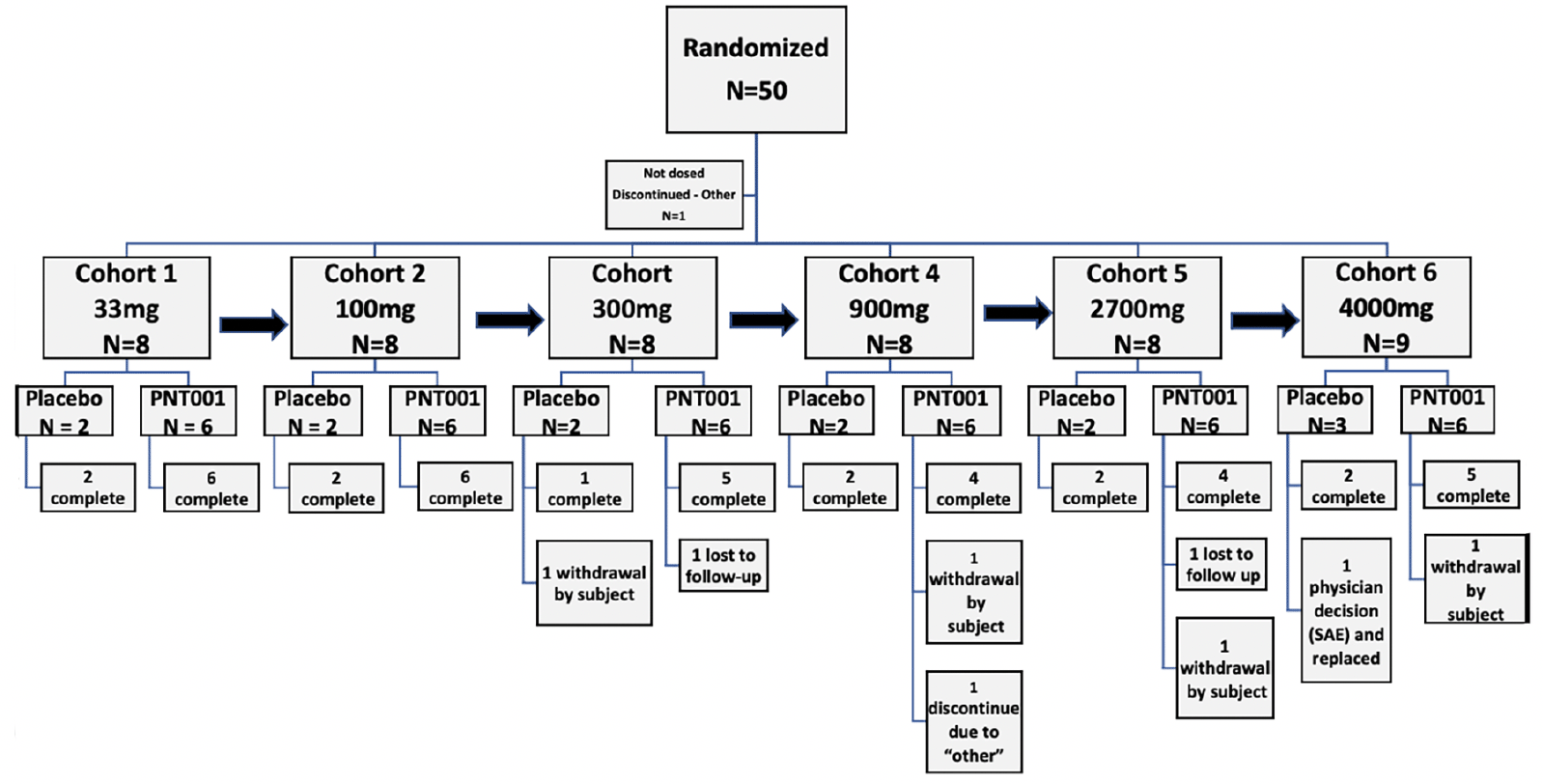
Figure 3. Enrollment and Disposition Diagram
The most reported system organ classes (SOCs) among all subjects were nervous system disorders and injury, poisoning and procedural complications with five subjects reporting at least one TEAE in each SOC. Three subjects (6.1%) reported at least one TEAE in both investigations and musculoskeletal and connective tissue disorders SOCs. No more than two subjects (4.1%) reported at least one event in all other SOCs. The most commonly reported preferred term (PT) was headache, with three subjects (6.1%) reporting at least one TEAE. All other PTs had no more than two subjects report at least one event. All TEAEs were rated as either mild (Grade 1; n = 19) or moderate (Grade 2; n = 16). The majority of TEAEs resolved with minimal intervention, six (17.1%) TEAEs were not resolved at the time of the subject’s study completion, and concomitant medication was used for 11 (31.4%) events. No unresolved TEAEs or those requiring medication were related to PNT001.
Of the 36 subjects in the PNT001 dose cohorts, three subjects (8.3%) reported one event each that was determined to be related to study treatment. The most reported SOCs in the PNT001 subjects were nervous system disorders, with four subjects (11.1%) reporting at least one TEAE, and injury, poisoning and procedural complications, with three subjects (8.3%) reporting at least one TEAE. Two subjects reported at least one event for each of the following PTs: headache, post-procedural complications, back pain, and procedural pain. All other PTs had no more than one subject reporting at least one TEAE. For eight of the 13 subjects (61.5%) in the placebo groups reporting at least one TEAE, injury, poisoning and procedural complications and respiratory, thoracic, and mediastinal disorders were the most reported SOCs with two subjects (15.4%) reporting at least one TEAE in each. No other SOC had reports by more than one subject, and no PT was reported by more than one subject.
Three (8.6%) of the 35 total events were determined to be related to the study drug. These were all Grade-1 events and occurred in the lowest dose cohort groups. In Cohort 1 (33 mg PNT001), two events were reported: a post-procedural complication (anxiety over lumbar puncture) and erythematous rash. One event of increased CSF protein was reported in Cohort 2 (100 mg PNT001). No TEAEs were reported in cohorts with PNT001 doses exceeding 100 mg.
Analysis of AEs
PNT001 was well tolerated with limited AEs overall and only three related events. A greater percentage of subjects receiving placebo experienced AEs than those in the PNT001 dose cohorts (61.5% versus 38.9%). All AEs were Grade 2 or less, and no more than two events occurred within the treatment group for any dose cohort. One placebo subject (2.0%) experienced one SAE of treatment noncompliance with concomitant medications that led to seizures. This SAE was not related to study treatment and resolved. There were no SAEs or AEs that led to early study termination or death (Table 2).
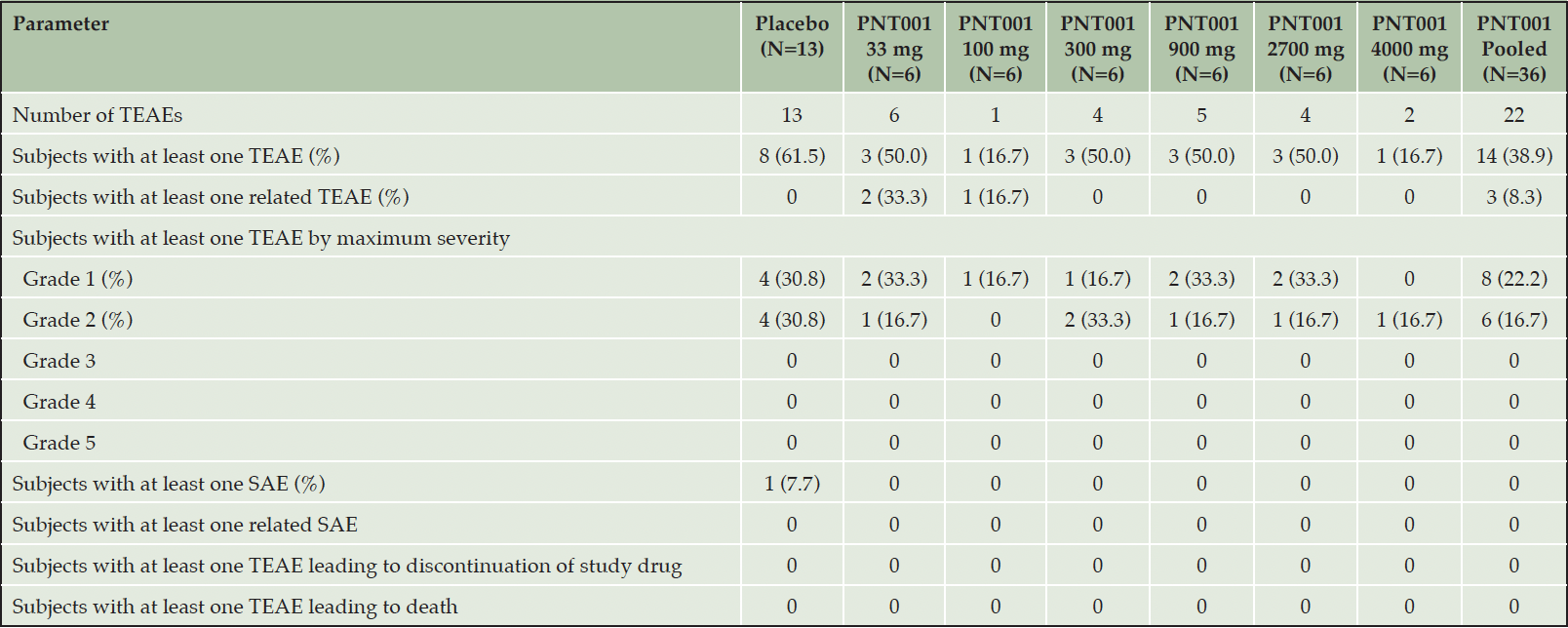
Table 2. Overview of Adverse Events by PNT001 in the Dose-Cohort Group (Safety Analysis Set)
Abbreviations: TEAE, treatment-emergent adverse event; SAE, serious adverse event.
SAEs
One SAE was reported in a subject who was randomized to and received placebo. The initial sentinel subject in Cohort 6 (4,000 mg) experienced a moderate, unrelated event of treatment noncompliance with concomitant medications that led to seizures after administration of the study drug. The subject was noncompliant with their prescribed concomitant medications and was found to have a 12-year history of seizures that had not been disclosed to study site staff when reviewing their medical history during the screening process. Medical records were requested and showed that, prior to entering the study, the subject was being switched from lacosamide (200 mg twice daily) to lamotrigine (150 mg twice daily). However, it was not clear if the subject took either medication as the subject had a long history of medication non-compliance. The blind was broken for this subject. This subject had received placebo and was replaced on the study per protocol due to ineligibility.
The AEs were relatively comparable between the placebo and treatment subjects. A greater percentage of placebo subjects (61.5%) experienced at least one AE compared to the treatment subjects (38.9%). The dose level of PNT001 administered to the subjects did not alter the safety profile. There were no indications of any negative effects on any laboratory parameters, vital signs, physical or neurological examinations, or suicidality risk due to the administration of PNT001 at any dose level. Similar findings were observed across all six dose cohorts. No suicidal ideation, behavior, actual attempts, or emergence of suicidality as assessed by the Columbia-Suicide Severity Rating Scale (C-SSRS) were found throughout the study (9). Overall, the safety data demonstrated a positive safety profile of PNT001 in healthy volunteers. There were no major safety concerns identified in this first-in-human study of PNT001.
Other Safety Results
Analyses of the clinical laboratory assessments, vital signs, physical and neurological examinations, electrocardiograms, and suicidality risk identified no remarkable safety concerns and no apparent differences between subjects that received PNT001 and those that received the same volume of placebo.
Anti-drug Antibody Results
Of the 36 subjects receiving PNT001, one post-baseline positive anti-drug antibody result was observed at Day 112 in a subject in Cohort 3 (300 mg PNT001). Three additional positive anti-drug antibody results were observed: one at baseline in a subject who was randomized to Cohort 6 (4,000 mg PNT001) and two in subjects randomized to placebo. Single-dose administration yielded a low rate of anti-drug antibodies, suggesting that PNT001 is not robustly immunogenic.
PK Results
As shown in Figure 1, following the intravenous infusion of PNT001 over approximately 30 minutes (dose levels 33–2,700 mg) or 60 minutes (4,000 mg), the median Tmax values of PNT001 in serum varied across the doses, ranging from 0.692 to 2.50 hours, after the start of dose infusion (individual Tmax values ranged from 0.517 to 8.97 hours after the start of infusion). After reaching Cmax, the serum concentrations of PNT001 declined in a generally biphasic manner. The geometric mean t1/2 values were similar across the dose levels, with values ranging from 23.8 to 33.8 days (Figure 1). For individual subjects, the t1/2 values ranged from 13.0 to 46.4 days across all dose levels and did not appear to be dose dependent. There were no clear trends in the geometric mean clearance (CL) or volume of distribution (Vd) across the doses (Figure 1).
Over the dose range of 33–4,000 mg, the geometric means of AUC0-∞, AUC0-t, and Cmax increased in a near dose proportional manner (the slopes for all three parameters were approximately 0.95, with the 95% CIs spanning unity); however, the assumption of linearity was unacceptable (lack of fit P-value was <0.05 for all three parameters). Thus, the power model was deemed not appropriate. The overall P-values for each of the dose-normalized AUC0-∞, AUC0-t, and Cmax values were ≤0.05 and considered statistically significant. Thus, an ANOVA model that included dose as a factor was used. A conclusion of dose proportionality could not be made over the evaluated dose range of 33–4,000 mg.
In general, the geometric coefficient of variation (CV) analyses showed that the between subject variabilities for AUC0-∞ and AUC0-t were low at the 33- and 100-mg dose levels (14.5% to 21.3%) but moderate to high at dose levels of 300 mg and higher (28.3% to 52.1%). The between-subject variability for Cmax was generally low with the geometric CV ranging from 11.7% to 23.4% over all dose levels except for 300 mg, for which the geometric CV was 45.5%.
The CSF concentrations of PNT001 at days 3 and 28 following dosing were dose proportional and virtually identical (Figure 2). The CSF concentration of PNT001 was approximately 0.1% of the total plasma concentration. These data indicate that single doses of PNT001 of 900 mg and above can achieve a sustained CSF concentration above the antigen-antibody Kd.
Discussion
The results of this first-in-human study of PNT001 showed a tolerable safety profile of PNT001 after a single dose in healthy volunteers over a dose range of 33–4,000 mg. Just under 35% of the subjects enrolled in the study were non-white, thus providing an assessment of PNT001 in a diverse population. There were no dose-limiting AEs and no withdrawals due to treatment-related AEs. The related AEs were mild and resolved without intervention. No maximum tolerated dose was identified.
Exposure was linear in the CSF following a single dose. The serum PK parameters were as expected for a monoclonal antibody. The PK data indicated that doses of PNT001 at and above 900 mg provided CSF concentrations of PNT001 above the dissociation constant (Kd) for PNT001 binding to cis-pT231 (45 ng/mL) and increased linearly with subsequent doses. The PK data indicated that therapeutic doses of PNT001, based on serum and CSF concentrations associated with improvement in the mouse model (7), were achievable within the dose range. The half-life results (23.8–33.8 days) suggest that monthly dosing in multiple dose studies may be appropriate. In addition, the high levels of PNT001 detected in CSF 28 days after dosing indicated that therapeutic levels of PNT001 may be maintained in the brain interstitial fluid with multiple dosing.
Passive immunization with anti-tau antibodies is designed to capture transmissible, pathologic tau species in the interstitial fluid between neurons (10-12). The results of independent studies from several laboratories have suggested that strains of pathologic tau exist across tauopathies (13-14).
The evolution of the understanding of the molecular composition of the transmissible species has driven a shift in focus from antibodies targeting the amino terminal of tau to antibodies like PNT001 that target the proline-rich region (PRR)/microtubule-binding region (MTBR) region.
The first clinically tested tau antibodies targeted the amino-terminal domain of tau. Gosuranemab, tilavonemab, and semorinemab all target the amino terminal domain. With a single exception (https://ir.acimmune.com/news-releases/news-release-details/ac-immune-announces-interim-phase-1b2a-data-showing-its-aci), these antibodies have not shown any benefits in clinical studies of patients with either PSP or early AD/Mild Cognitive Impairment (15-17). The clinical development of zagotenemab has been discontinued due to lack of efficacy (18). The epitope for this antibody comprises both an amino terminal segment and an MTBR segment.
It is unclear whether these results are due to a low abundance of the amino terminus in transmissible tau species, as has been reported in CSF (19-22), inaccessibility of this region for antibody binding(23–28), or intervention that is too late (29) in disease progression. Nevertheless, there may be shared epitopes among the different tau strains, such as the PRR- and microtubule-binding regions (30) that are targeted by PNT001 and antibodies currently in PhII (31-37), as well as pan-tau approaches, such as aggregation inhibitors or antisense oligomers that are in or close to clinical development (38).
The supportive pre-clinical data and safety of single doses at potentially therapeutic dose levels indicate the feasibility of multiple-dose patient studies of PNT001 in tauopathies.
Funding: This work was supported by Pinteon Therapeutics, Inc.
Acknowledgements: We thank Dr. Larry Altstiel for his assistance with manuscript preparation and data analysis. We would also like to thank the subjects who participated in the trial and the study site staff who supported the trial during the pandemic.
Conflict of interest: WL, KF, KM, MJ, and MA are or were employees of Pinteon Therapeutics, Inc. As such, each received both cash compensation and stock options.
Ethical standards: This study was conducted in accordance with the protocol, applicable regulatory requirements, International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Conference on Harmonisation Harmonised Tripartite Guideline (E6), «Guideline for Good Clinical Practice», the ethical principles that have their own origin in the principles of the Declaration of Helsinki, as well as Title 21 of the United States Code of Federal Regulations, Parts 50, 56, and 312.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. Nakamura K, Greenwood A, Binder L, et al. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer’s disease. Cell 2012;149:232-244. https://doi.org/10.1016/j.cell.2012.02.016.
2. Kondo A, Shahpasand K, Mannix R, et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 2015;523:431-436. https://doi.org/10.1038/nature14658.
3. Lu KP, Kondo A, Albayram O, Herbert MK, Liu H, Zhou XZ. Potential of the antibody against cis-phosphorylated tau in the early diagnosis, treatment, and prevention of Alzheimer Disease and brain injury. JAMA Neurol 2016;73:1356-1362. https://doi.org/10.1001/jamaneurol.2016.2027.
4. Albayram O, Herbert MK, Kondo A, et al. Function and regulation of tau conformations in the development and treatment of traumatic brain injury and neurodegeneration. Cell Biosci 2016;6:59. https://doi.org/10.1186/s13578-016-0124-4.
5. Albayram O, Kondo A, Mannix R, et al. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun 2017;8:1000. https://doi.org/10.1038/s41467-017-01068-4.
6. Qiu C, Albayram O, Kondo A, et al. Cis P-tau underlies vascular contribution to cognitive impairment and dementia and can be effectively targeted by immunotherapy in mice. Sci Transl Med 2021;13:eaaz7615. https://doi.org/10.1126/scitranslmed.aaz7615.
7. Foster K, Manca M, McClure K, et al. Preclinical characterization and IND-enabling safety studies for PNT001, an antibody that recognizes cis-pT231 tau. Alzheimer’s Dement 2023. https://doi.org/10.1002/alz.13028.
8. US Department of Health and Human Services, Food and Drug Administration. Guidance for Industry Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. 2005. Rockville, MD.
9. Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry 2011;168:1266-1277. https://doi.org/10.1176/appi.ajp2011.10111704.
10. Brettschneider J, Del Tredici K, Lee VMY, Trojanowski JQ. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci 2015;16:109-120. https://doi.org/10.1038/nrn3887.
11. Colin M, Dujardin S, Schraen-Maschke S, et al. From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy. Acta Neuropathol 2020;139:3-25. https://doi.org/10.1007/s00401-019-02087-9.
12. Gibbons GS, Lee VMY, Trojanowski JQ. Mechanisms of cell-to-cell transmission of pathological tau: a review. JAMA Neurol 2019;76:101-108. https://doi.org/10.1001/jamaneurol.2018.2505.
13. Sanders DW, Kaufman SK, DeVos SL, et al. Distinct Tau Prion Strains Propagate in Cells and Mice and Define Different Tauopathies. Neuron 2014;82:1271-1288. https://doi.org/10.1016/j.neuron.2014.04.047.
14. Kaufman SK, Sanders DW, Thomas TL, et al. Tau Prion Strains Dictate Patterns of Cell Pathology, Progression Rate, and Regional Vulnerability In Vivo. Neuron 2016;92:796-812. https://doi.org/10.1016/j.neuron.2016.09.055.
15. Dam T, Boxer AL, Golbe LI, et al. Safety and efficacy of anti-tau monoclonal antibody gosuranemab in progressive supranuclear palsy: a phase 2, randomized, placebo-controlled trial. Nat Med. 2021;27:1451-1457. https://doi.org/10.1038/s41591-021-01455-x.
16. Höglinger GU, Litvan I, Mendonca N, et al. Safety and efficacy of tilavonemab in progressive supranuclear palsy: a phase 2, randomised, placebo-controlled trial. Lancet Neurol 2021;20:182-192. https://doi.org/10.1016/S1474-4422(20)30489-0.
17. Jabbari E, Duff KE. Tau-targeting antibody therapies: too late, wrong epitope or wrong target? Nat Med 2021;27:1341-1342. https://doi.org/10.1038/s41591-021-01465-9.
18. Mullard A. Anti-tau antibody failures stack up. Nat Rev Drug Discov 2021;20:888. https://doi.org/10.1038/d41573-021-00187-4.
19. Barthelemy NR, Fenaille F, Hirtz C, et al. Tau protein quantification in human cerebrospinal fluid by targeted mass spectrometry at high sequence coverage provides insights into its primary structure heterogeneity. J Proteome Res 2016. https://doi.org/10.1021/acs.jproteome.5b01001.
20. Barthelemy NR, Gabelle A, Hirtz C, et al. Differential mass spectrometry profiles of Tau protein in the cerebrospinal fluid of patients with Alzheimer’s disease, progressive supranuclear palsy, and dementia with Lewy Bodies. J Alzheimers Dis 2016;51:1033-1043. https://doi.org/10.3233/JAD-150962.
21. Horie K, Barthélemy NR, Sato C, Bateman RJ. CSF tau microtubule binding region identifies tau tangle and clinical stages of Alzheimer’s disease. Brain 2021:144:515-527. https://doi.org/10.1093/brain/awaa373.
22. Meredith JE Jr, Sankaranarayanan S, Guss V, et al. Characterization of novel CSF Tau and ptau biomarkers for Alzheimer’s disease. PLoS One 2013;8:e76523. https://doi.org/10.1371/journal.pone.0076523.
23. Fitzpatrick AWP, Falcon B, He S, et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017;547:185-190. https://doi.org/10.1038/nature23002.
24. Falcon B, Zhang W, Murzin AG, et al. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018;561:137-140. https://doi.org/10.1038/s41586-018-0454-y.
25. Falcon B, Zivanov J, Zhang W, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019;568:420-423. https://doi.org/10.1038/s41586-019-1026-5.
26. Zhang W, Tarutani A, Newell KL, et al. Novel tau filament fold in corticobasal degeneration. Nature 2020;580:283-287. https://doi.org/10.1038/s41586-020-2043-0.
27. Shi Y, Murzin AG, Falcon B, et al. Cryo-EM structures of tau filaments from Alzheimer’s disease with PET ligand APN-1607. Acta Neuropathol 2021;141: 697-708. https://doi.org/10.1007/s00401-021-02294-3.
28. Goedert M. Cryo-EM structures of tau filaments from human brain. Essays Biochem 2021;65: 949-959. https://doi.org/10.1042/EBC20210025.
29. Meisl G, Hidari E, Allinson K, et al. In vivo rate-determining steps of tau seed accumulation in Alzheimer’s disease. Sci Adv 2021;7:eabh1448. https://doi.org/10.1126/sciadv.abh1448.
30. Wesseling H, Mair W, Kumar M, et al. Tau PTM profiles identify patient heterogeneity and stages of Alzheimer’s disease. Cell 2020;183:1699-1713.e13. https://doi.org/10.1016/j.cell.2020.10.029.
31. Courade JP, Angers R, Mairet-Coello G, et al. Epitope determines efficacy of therapeutic anti-Tau antibodies in a functional assay with human Alzheimer Tau. Acta Neuropathol 2018;136: 729-745. https://doi.org/10.1007/s00401-018-1911-2.
32. Albert M, Mairet-Coello G, Danis C, et al. Prevention of tau seeding and propagation by immunotherapy with a central tau epitope antibody. Brain 2019;142:1736-1750. https://doi.org/10.1093/brain/awz100.
33. Van Kolen K, Malia TJ, Theunis C, et al. Discovery and functional characterization of hPT3, a humanized anti-phospho Tau selective monoclonal antibody. J Alzheimers Dis. 2020;77:1397-1416. https://doi.org/10.3233/JAD-200544.
34. Roberts M, Sevastou I, Imaizumi Y, et al. Pre-clinical characterisation of E2814, a high-affinity antibody targeting the microtubule-binding repeat domain of tau for passive immunotherapy in Alzheimer’s disease. Acta Neuropathol Commun. 2020;8:13. https://doi.org/10.1186/s40478-020-0884-2.
35. d’Abramo C, Acker CM, Jimenez HT, Davies P. Tau passive immunotherapy in mutant P301L mice: antibody affinity versus specificity. PLoS One 2013;8:e62402. https://doi.org/10.1371/journal.pone.0062402.
36. Ji C, Sigurdsson EM. Current status of clinical trials on Tau immunotherapies. Drugs 2021;81: 1135-1152. https://doi.org/10.1007/s40265-021-01546-6.
37. Imbimbo BP, Ippati S, Watling M, Balducci C. A critical appraisal of tau-targeting therapies for primary and secondary tauopathies. Alzheimers Dement 2022;18:1008-1037. https://doi.org/10.1002/alz.12453.
38. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of IONIS-MAPTRx in Patients With Mild Alzheimer’s Disease. ClinicalTrials.gov identifier: NCT03186989. Last updated: 27 February 2023. https://ClinicalTrials.gov/show/NCT03186989. Accessed 7 September 2023.
© The Authors 2024