
M. Owen1, N. Bose2, L. Nisenbaum1, K.A. Partrick3, H.M. Fillit1
1. Alzheimer’s Drug Discovery Foundation, 57 West 57th St. Suite 904, New York, NY 10019, USA; 2. Gates Ventures, 4110 Carillon Point, Kirkland, WA 98033, USA; 3. Independent Researcher, 87 Providence Dr., Asheville, NC 27409, USA
Corresponding Author: Howard M Fillit, MD, Alzheimer’s Drug Discovery Foundation, 57 West 57th St. Suite 904, New York, NY 10019, USA, Email: hfillit@alzdiscovery.org, Phone: 1-212-901-8000
J Prev Alz Dis 2023;4(10):729-742
Published online October 24, 2023, http://dx.doi.org/10.14283/jpad.2023.111
Abstract
Alzheimer’s disease is a devastating neurodegenerative disorder that poses a significant societal burden. Approval of anti-amyloid antibody therapies is a significant milestone for treatment that was enabled by the inclusion of biomarkers. The use of biomarkers in clinical trials for Alzheimer’s disease has enabled selective participant recruitment, improved treatment monitoring, and supported more rigorous trial designs. This review discusses emerging biomarkers associated with the biology of aging and their application in Alzheimer’s disease clinical trials. Aging is the primary risk factor for sporadic Alzheimer’s disease and is associated with biological processes implicated in disease development and progression. Novel therapies targeting these underlying biological aging processes are currently undergoing clinical development. Biomarkers that capture the biology of aging are integral to accelerating the development of these therapies. Current progress in biomarker development demonstrates efforts to capture the full spectrum of aging biology. Further work is needed to expand the range of biomarkers that enable comprehensive assessment of brain pathology and aid in prognosis, diagnosis, and measuring treatment response. Establishing a comprehensive arsenal of biomarkers will support strategic decision making and increase the likelihood of positive clinical trials and drug registration for the next generation of Alzheimer’s disease drugs targeting the biology of aging.
Key words: Alzheimer’s disease, biomarkers, aging biology, clinical trials, drug development.
Introduction
Alzheimer’s disease (AD), the leading cause of dementia, is characterized by accumulation of abnormal beta-amyloid proteins (Aβ), phosphorylated tau (p-tau), and neuronal degeneration with subsequent cognitive impairment. An estimated 6.7 million Americans are currently living with Alzheimer’s dementia, or 1 in 9 people over 65 (1). The Food and Drug Administration (FDA) has approved seven medications for the treatment of AD: five aimed at improving symptoms and two anti-amyloid antibodies, aducanumab and lecanemab, that remove Aβ from the brain and slow cognitive decline.
A critical component of the recent success of clinical trials testing anti-amyloid antibody therapies was the inclusion of and reliance on biomarkers of AD pathology. Prior to 2012, an accurate diagnosis of AD required a postmortem examination of the brain to confirm the presence of amyloid plaques. Because early drug trials in AD relied heavily on clinical diagnosis, it was later estimated that up to one third of patients enrolled may not have had measurable amyloid in their brains (2, 3). The development of validated biomarkers of amyloid plaque burden, such as positron emission tomography (PET) and cerebrospinal fluid (CSF) assays, played a central role in the accelerated approval of aducanumab (4) and lecanemab (5). The inclusion of amyloid biomarkers in clinical trials of anti-amyloid antibody therapies enabled greater rigor by facilitating accurate diagnosis, supporting appropriate patient selection, demonstrating evidence of target engagement, and quantifying pharmacodynamic drug effects. While the approval of anti-amyloid antibodies is a promising advance for treating AD, these treatments are currently only approved for use in early disease, require bi-weekly or monthly intravenous infusions, suffer from potentially serious side effects that require consistent monitoring, and slow decline by approximately 27% (5). Additional therapeutic options are needed to treat and prevent AD.
The etiology of AD is complex with a mixture of risk factors including family history, genetics, education, brain injury, and age. Other chronic age-related diseases, such as cancer and hypertension, have seen improved outcomes from a combination therapy approach. Like these age-related diseases, common biological aging processes are implicated in the development and progression of AD (6) including inflammation, senescence, vascular dysfunction, aberrant proteostasis, synaptic dysfunction, mitochondrial oxidative stress, metabolic dysfunction including insulin resistance, and epigenetic changes. Because aging is the leading risk factor for sporadic AD, the biology of aging offers a promising framework upon which to pursue a combination therapy approach for dementia (7). To achieve this, a diverse set of biomarkers is needed to support the development of drugs that target the biology of aging.
Novel drugs targeting the biology of aging are already increasingly represented in the current pipeline of AD drugs in development. According to a 2023 report, within all clinical trial phases, agents are targeting amyloid (n=22, 16%) and tau (n=13, 9%), but also inflammation (n=24, 17%), senescence (n=1, <1%), vasculature (n=2, 1%), proteostasis (n=4, 3%), synaptic plasticity/neuroprotection (n=18, 13%), oxidative stress (n=7, 5%), metabolism and bioenergetics (n=10, 7%), and epigenetics (n=1, <1%) (8). For these trials, biomarkers of amyloid may be used for diagnostic confirmation, but other markers will be important for patient selection, monitoring, pharmacodynamic measurement, and response prediction. This review discusses emerging biomarkers associated with the biology of aging and their application in AD clinical trials.
Progress is being made in developing a broader arsenal of biomarkers to capture the full spectrum of the biology of aging using imaging, fluid, retinal, and digital tools; however, efforts must continue to discover, develop, and validate these biomarkers to support strategic decision making and increase likelihood of positive clinical trials and drug registration for the next generation of AD drugs. Successfully implementing a more robust biomarker-therapeutic strategy in clinical trials could truly enable precision medicine and combination therapies for AD.
The impact of biomarkers in clinical trials for Alzheimer’s disease
The FDA defines a biomarker as an indicator of normal biological processes, pathogenic processes, or biological responses to an exposure or intervention, including a therapeutic intervention (9). Significant resources have been devoted to developing and validating biomarkers that measure the underlying pathology of AD. PET imaging, CSF assays, and blood tests are now available to measure pathological hallmarks of the disease, including amyloid and tau. Additionally, noninvasive tools such as retinal scans and the analysis of digital signatures have begun to emerge and show promise.
To best support clinical trials, biomarkers must have a defined context of use (COU) and supportive data for that COU. In the Biomarkers, Endpoints and Other Tools (BEST) guidelines, the FDA defines COU as the biomarker’s intended use in drug development as either a risk/susceptibility, diagnostic, monitoring, predictive, prognostic, pharmacodynamic, or safety biomarker (9). Clinical trials using biomarkers have a greater success rate than those that do not (10), particularly when used strategically in multiple COUs (11, 12). For central nervous system (CNS) diseases, biomarkers are needed to assess a drug’s ability to cross the blood-brain barrier (BBB), such as an assay that can directly measure the drug in CSF. Biomarkers can also help ensure the enrollment of the right patients at the right time. Patients can be screened for Alzheimer’s pathology using biomarkers of amyloid, tau, and neurodegeneration (13) but also for the specific pathology targeted by the drug’s mechanism of action. Moreover, biomarkers can be used to show evidence that the drug engages the target, or target engagement, a feature that is particularly important for Phase 2 clinical trials (14). Target engagement can be shown for drugs targeting a specific receptor in Phase 0 studies that predict receptor occupancy, regional distribution, and brain penetration using PET (15). Additionally, biomarkers can show proof of pharmacology or more downstream biological effects of successful target engagement. An effective biomarker strategy should assess whether there are appropriate biomarkers and relevant assays that accompany each COU as needed.
Emerging biomarkers for the biology of aging
An expanded arsenal of available, robust, and validated biomarkers for multiple COUs is needed for AD clinical trials investigating drugs targeting the biology of aging. Table 1 provides examples of emerging imaging and fluid biomarkers for the biology of aging that are under investigation to determine their utility in AD clinical trials. Notably, while some biomarkers are available and others are currently in development, they remain lacking for many drug targets.
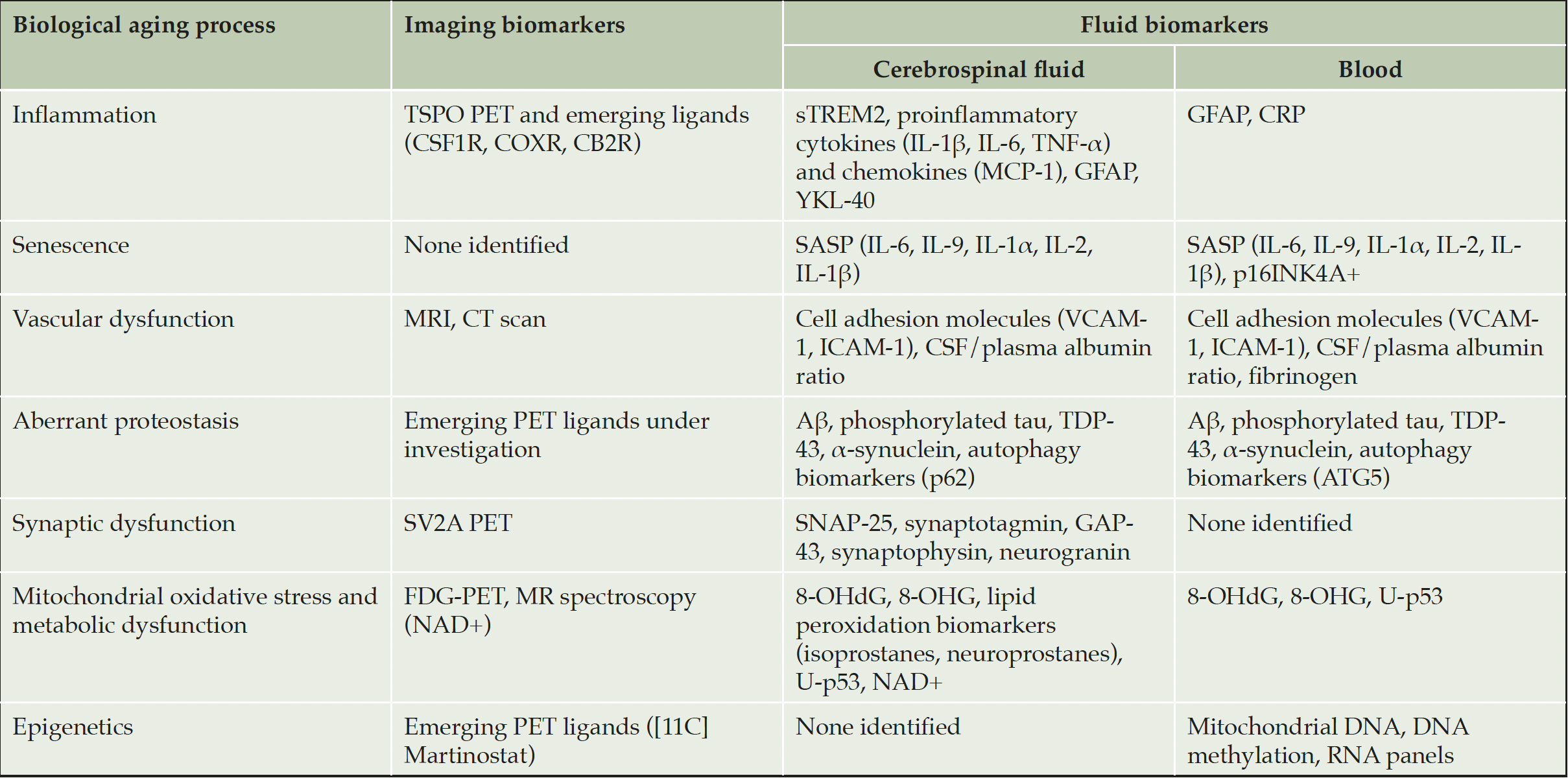
Table 1. Emerging biomarkers for the biology of aging
Table 1 provides examples of emerging biomarkers that may be considered for use in clinical trials for drugs targeting biological aging processes implicated in AD and is not an exhaustive list of all relevant biomarkers. Many of these biomarkers require further investigation to fully understand their potential utility in clinical trials. 8-OHdG = 8-hydroxy-2’deoxyguanosine; 8-OHG = 8-hydroxy-2’oxyguanisine; Aβ = beta-amyloid protein; ATG5 = autophagy-related gene 5; CB2R = Cannabinoid receptor type 2; COXR = cyclooxygenase receptor; CRP = c-reactive protein; CSF = cerebrospinal fluid; CSF1R = colony-stimulating factor 1 receptor; CT = computerized tomography; DNA = deoxyribonucleic acid; FDG = fluorodeoxyglucose; GAP-43 = growth associated protein 43; GFAP = glial fibrillary acidic protein; IL = interleukin; ICAM-1 = intercellular adhesion molecule-1; MCP-1 = monocyte chemoattractant protein-1; MR = magnetic resonance; MRI = magnetic resonance imaging; NAD+ = nicotinamide adenine dinucleotide; PET = positron emission tomography; RNA = ribonucleic acid; SASP = senescence-associated secretory phenotype; SNAP-25 = synaptosomal-associated protein of 25 kDa; sTREM2 = soluble triggering receptor expressed on myeloid cells 2; SV2A = synaptic vesicle glycoprotein 2A; TDP-43 = TAR DNA binding protein-43; TNF-α = tumor necrosis factor alpha; TSPO = translocator protein-18 kDa; U-p53 = unfolded conformational variant of p53; VCAM-1 = vascular cell adhesion molecule-1.
Inflammation
Inflammation is a hallmark of aging and significant contributor to age-related cognitive decline and dementia risk (7, 16). While broad-spectrum anti-inflammatory drugs have largely failed in trials for AD, those targeting specific aspects of inflammation while sparing others show promise. Twenty-four anti-inflammatory agents are currently in the pipeline (8). As novel therapies targeting inflammation continue to enter clinical development, identification and validation of biomarkers targeting specific inflammatory pathways will be necessary for selecting promising drug candidates and expediting their development.
Microglia and astrocytes are key regulators of neuroinflammation and play central roles in the development and progression of AD (17). Microglia are the primary innate immune cells in the brain that release proinflammatory mediators in response to pathogens and CNS insults and support tissue repair. They also protect the brain by inducing phagocytosis of apoptotic cells and other neuronal debris (18). The role of microglia in AD is complex and differs based on the cell’s state and function. In addition, microglial function evolves throughout disease progression. Depending on the phenotype, activated microglia can exacerbate AD pathology or provide protective effects (19, 20). The robust proinflammatory cascade induced by microglia increases susceptibility for AD and contributes to disease progression, in part by accelerating neurodegeneration (19). However, activated microglia can also reduce AD pathology by phagocytosis of amyloid plaques (21). Biomarkers are needed that reflect the complexity of microglia in AD to better understand microglia phenotypes and how they change over time. Further, biomarkers for microglia subtypes could accelerate the development of more strategic, targeted approaches that reduce chronic neuroinflammation while sparing or promoting the phagocytosis of amyloid plaque and other neuronal debris.
Translocator protein-18 kDa (TSPO) PET is an imaging biomarker of neuroinflammation that is currently being used as a pharmacodynamic biomarker as well as a monitoring biomarker to evaluate the effect of anti-inflammatory therapies. TSPO PET is being employed as a secondary outcome measure in Phase 1 clinical studies of anti-inflammatory agents for mild cognitive impairment (MCI) or early AD (NCT04795466, NCT05468073). Although TSPO is commonly used as a marker of inflammatory glia, it is not specific to this population of glia, or even to microglia more broadly, as it is also expressed on endothelial cells (22). Markers that demonstrate higher specificity for microglia activation, such as colony-stimulating factor 1 receptor, cyclooxygenase receptor, and Cannabinoid receptor type 2, are currently under investigation (23).
Triggering receptor expressed on myeloid cells 2 (TREM2) is a lipid receptor expressed in microglia (24). Mutations in TREM2 increase the risk of AD (25, 26), and reduction of TREM2 activity has been shown to reduce microglial phagocytosis of amyloid plaques (27). Thus, drugs that improve TREM2 signaling may provide therapeutic value in AD by enhancing microglia activity to increase clearance of amyloid pathology. sTREM2, a soluble variant of TREM2, has been developed as a fluid biomarker of microglia activity (28). CSF sTREM2 was used as a target engagement biomarker in a Phase 1 study of AL002, a monoclonal Immunoglobulin G1 antibody therapy targeting TREM2, where a dose-dependent reduction in sTREM2 indicated successful target engagement. This study also evaluated soluble colony-stimulating factor 1 receptor (sCSF1R), secreted phosphoprotein 1 (SPP1), and interleukin-1 receptor antagonist (IL1RN) (29), biomarkers of microglia function that occur downstream of TREM2 signaling. AL002 is currently in Phase 2 development for early AD and change from baseline in CSF sTREM2, sCSF1R, SPP1, IL1RN will be assessed as pharmacodynamic biomarker endpoints (NCT04592874).
Many of the proinflammatory mediators that comprise microglia-induced neuroinflammation have been suggested as fluid biomarkers of microglia activity. Proinflammatory cytokines such as interleukin (IL)-1β, IL-6, and tumor necrosis factor alpha (TNF-α) initiate and sustain complex interconnected pathways that drive neuroinflammation in AD (19). CSF IL-1β, IL-6, TNF-α biomarkers are currently being used as primary or secondary outcome measures in early phase clinical trials to assess the treatment effect of novel anti-inflammatory therapies in early (NCT04804241, NCT04381468) or general AD (NCT03943264).
Proinflammatory chemokines also play an important role in neuroinflammation and offer additional biomarker targets. Monocyte chemoattractant protein-1 (MCP-1) is a proinflammatory chemokine involved in the regulation and recruitment of immune cells to sites of inflammation (30, 31). CSF MCP-1 levels are elevated in MCI and AD and correlate with cognitive decline across the disease spectrum (32), suggesting its use as a monitoring biomarker of disease progression. In an ongoing Phase 1/2 trial of the repurposed therapy baricitinib, CSF MCP-1 will be used as a predictive biomarker in patient selection as well as a pharmacodynamic biomarker to evaluate baricitinib’s effects in individuals with AD (NCT05189106). CSF MCP-1 is also being assessed, in conjunction with proinflammatory cytokines, as a primary outcome measure in a Phase 2 study of senicapoc, a novel disease-modifying therapy for early AD (NCT04804241). Fluid biomarkers targeting proinflammatory cytokines and chemokines provide information on cerebral inflammatory activity and can be leveraged in clinical trials to help identify the appropriate patient as well as for pharmacodynamic assessment and monitoring of inflammation status.
Astrocytes are a subtype of glial cells that are critical to the health and function of neurons, contributing to processes such as promoting synapse formation and modulating neurotransmission (33). Like microglia, astrocytes can be activated in ways that exert detrimental or beneficial functions, commonly referred to as type A1 and A2, respectively (34, 35). With age, astrocytes become reactive through a transition termed astrogliosis (36). Reactive astrocytes can contribute to neuroinflammation, and like microglia, have been observed to cluster around amyloid plaques (19). Reactive astrocytes are characterized by increased expression of glial fibrillary acidic protein (GFAP) (37). GFAP is a sensitive biomarker for detecting and monitoring astrogliosis throughout disease progression (32, 38) and could serve as a target engagement biomarker for astrocyte-targeting therapies. Further, elevated plasma GFAP is associated with Aβ elevation and predicts cognitive decline and conversion to AD in both cognitively unimpaired individuals and those with MCI (39, 40), demonstrating value as a prognostic biomarker. Based on this connection with Aβ, GFAP has also shown promise as a pharmacodynamic and monitoring biomarker in clinical trials of the anti-amyloid antibodies donanemab (41) and lecanemab (42). However, GFAP does not reliably differentiate between A1 and A2 astrocytes. A1 astrocytes are associated with an increase in proinflammatory molecules such as C1q, TNFα, and IL-1α, while A2 astrocytes are associated with a reduction in inflammatory cytokines, such as IL-6, and IL-1β, but changes in levels of these circulating cytokines are generally non-specific (34). Novel markers are needed to monitor the change in the profile of neuroinflammatory and neuroprotective astrocytes in response to immunomodulatory therapies.
YKL-40 is a glycoprotein expressed in both reactive astrocytes and activated microglia (19, 43). YKL-40 expression levels are significantly higher in individuals with MCI due to AD compared to those with MCI due to other causes (44) and can differentiate AD from other neurodegenerative diseases (32). These data suggest YKL-40 is a potential biomarker to support a differential diagnosis, prognosis of disease progression, and target engagement for anti-inflammatory agents.
C-reactive protein (CRP) is a proinflammatory signaling protein released by hepatocytes in response to circulating inflammatory cytokines (45). Pathological studies have revealed the presence of CRP in amyloid plaques and tau tangles in AD brain tissue (46). CRP is being used as a pharmacodynamic biomarker in two Phase 3 trials of semaglutide, a repurposed therapy shown to have anti-inflammatory effects, in part by reducing CRP signaling (NCT04777396, NCT04777409) (47). Further, recent studies have identified CRP as a potential prognostic biomarker for conversion from MCI to AD (46) and risk biomarker in APOE ε4 allele carriers (48), suggesting its potential value as an inflammatory biomarker for multiple COUs.
Much progress has been made in advancing inflammatory biomarkers, resulting in a diverse set of markers to aid current and future clinical trial designs. Validation of imaging biomarkers more specific to microglia activation are currently underway (23). Research efforts should seek to identify additional imaging biomarkers that target different functions of microglia, as well as biomarkers that are specific to astrogliosis and other neuroinflammatory processes implicated in AD. To date, imaging and fluid inflammation biomarkers are mostly used as pharmacodynamic or monitoring biomarkers and some, such as GFAP and CRP, have shown promise as prognostic or risk biomarkers. Thus, a gap exists to validate biomarkers for other COUs, such as predictive biomarkers that help determine who may be most responsive to a drug. To improve testing of anti-inflammatory therapies for AD, a more comprehensive reserve of biomarkers that span different COUs as well as biomarkers that are specific to the mechanism of action of novel drug candidates are needed.
Senescence
Cellular senescence is characterized by cells that evade death and release proinflammatory cytokines and chemokines that induce tissue damage in the brain and other organs. Senescent cells accumulate with age and are implicated in AD (49). Senolytic therapies are beginning to be tested for use in AD (NCT04685590). However, the lack of specific, reliable biomarkers for target engagement and monitoring efficacy poses a challenge. Studies have revealed unique senescence transcriptomes by cell type and stressor type, emphasizing the need to further characterize senescent cells across different tissues and cells in the human body to advance biomarker studies and clinical trials (50).
Preclinical and clinical characterization of cellular senescence in AD has provided evidence for promising biomarkers to support clinical trials (49, 51, 52). Senescent cells secrete a variety of proinflammatory molecules, collectively known as the senescence-associated secretory phenotype (SASP), which contribute to inflammation, tissue degradation, and age-related diseases (49). Several SASP factors, IL-6, IL-9, IL-1α, IL-2, and IL-1β, are detected in the blood and CSF of individuals with AD, suggesting their potential as biomarkers for monitoring senescence. Further research is needed to improve the specificity of these biomarkers to senescence and distinguish them from other inflammatory responses. Additionally, some SASP factors detected in CSF may originate from peripheral sources, as peripheral factors can cross the BBB through bidirectional transport systems. Thus, considerations should be made to evaluate target engagement of new therapeutics in the periphery as well as the brain (49).
p16INK4a, a cell cycle regulator, is commonly used as a biomarker for cellular senescence. Its expression levels are known to increase in senescent cells (52, 53), and reduction in p16INK4a expression can indicate the effectiveness of senolytic interventions. Both SASP (composed of IL-6, IL-9, IL-1α, IL-2, IL-1β) and p16INK4A+ blood biomarkers are currently being used as secondary outcome measures in a Phase 2 clinical trial that evaluates the safety and efficacy of the combination of two senolytic therapies, dasatinib and quercetin, in older adults with biomarker-confirmed MCI or early-stage AD (NCT04685590) (51).
Additionally, exosomes, small extracellular vesicles that carry cargo of various biomolecules, are being explored for their potential as biomarkers of senescence in AD. Senescent cells release exosomes with unique biomolecule compositions, which provide information about the effects of senescence on specific cell types. Isolation and analysis of exosomes from the blood or other biofluids may shed light on the biology of senescence in AD and its contribution to disease progression. However, further research is needed to better understand the role of exosomes secreted by senescent cells in AD pathogenesis prior to their use in clinical trials (49). The search for reliable biomarkers of cellular senescence in AD is ongoing. Continued efforts should be employed to further characterize cellular senescence in AD and identify and validate novel biomarkers to improve the rigor of future clinical trials.
Vascular dysfunction
Vascular pathology occurs with aging and is a significant contributor to cognitive decline and the development of dementias, including AD (7, 54). A post-mortem study estimated that approximately 80% of people with AD have vascular pathology (55). Extensive research has shown that vascular impairment leads to reduced cerebral blood flow, endothelial damage, BBB leakage, demyelination, and cerebral atrophy, all of which can contribute to AD pathology and clinical decline (56, 57).
Neuroimaging has been the predominant biomarker used for vascular pathology in AD, thus far. Magnetic resonance imaging (MRI) is used to assess multiple aspects of vascular damage including abnormal cerebral blood flow, BBB leakage, microbleeds, and white matter hyperintensities (WMH) (58). Novel approaches are in development that use quantitative water permeability mapping to quantify BBB dysfunction (59). Computerized tomography scans allow the identification of vascular calcifications in the hippocampus, which have been linked to increased cognitive impairment (60).
Fluid biomarkers, including cell adhesion molecules, CSF/plasma albumin ratio, and fibrinogen, show promise in assessing vascular pathology in AD. Elevated levels of vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) are observed in the CSF and plasma from preclinical AD through later stages of AD dementia. These molecules are associated with subsequent cognitive impairment in individuals without dementia and have the potential to serve as markers for risk assessment and disease progression (61). The CSF/plasma albumin ratio reflects BBB integrity, with abnormal ratios indicating BBB damage. Although the relationship between this ratio and AD is not fully understood, it holds promise as a biomarker for differentiating AD from other disorders, in addition to its role in assessing BBB integrity (62). Fibrinogen, a glycoprotein involved in coagulation and vascular reactivity, can serve as a plasma biomarker for vascular damage (63). Fibrinogen interacts with amyloid, exacerbating clotting, fibrin deposition, and triggering proinflammatory signaling (64-66). Further, fibrinogen converts to fibrin through thrombin-mediated cleavage (67). Accumulation of fibrin deposits is observed in AD and correlates with pathology (68). Ongoing investigations are exploring fibrin deposits as an imaging biomarker for vascular damage and as a potential novel drug target for AD.
Of the biomarkers for vascular dysfunction, MRI is most often leveraged in clinical trials to monitor treatment efficacy and assess changes in disease progression. In a clinical study of the antiplatelet agent cilostazol, MRI was employed as a predictive marker to aid in patient selection. MRI also served as a safety biomarker in this study, measuring WMH severity to identify individuals who could safely be administered the drug (NCT01409564). In a Phase 2/3 study investigating eicosapentaenoic acid in cognitively unimpaired veterans at risk for AD, cerebral blood flow will be assessed using arterial spin-labeling MRI. Eicosapentaenoic acid is believed to reduce the risk of AD, in part by enhancing cerebral blood flow (NCT02719327). Additional efforts are needed to expand the use of biomarkers for vascular dysfunction.
Aberrant proteostasis
While Alzheimer’s pathology is predominantly characterized by the accumulation of abnormal Aβ proteins and phosphorylated tau, it is now more recognized that other misfolded or aggregated proteins are often present. TAR DNA binding protein-43 (TDP-43) pathology is detected in up to 52% of AD brains (69) where it has been associated with more severe atrophy and greater memory loss (70). Additionally, the presynaptic protein α-synuclein, mainly associated with Parkinson’s disease (PD), dementia with Lewy bodies, and multiple system atrophy, is present in over half of AD patients (71). The mean age for onset of dementia and death is younger for AD patients with Lewy bodies than for AD patients without Lewy bodies (72). Buildup of multiple pathological proteins suggests a potential breakdown in mechanisms involved with protein homeostasis or proteostasis.
To maintain normal proteostasis, neurons depend heavily on autophagy, a lysosomal-dependent degradation process, to clear misfolded protein aggregates (73). Neurons are especially vulnerable to autophagic dysfunction because lysosomes are concentrated in the cell body and autophagosomes have to be transported from distal axons to the cell body to have their contents degraded by these lysosomes (74, 75). Impaired autophagy is associated with aging (76) and AD (77). Autophagy modulates Aβ clearance (78) and tau degradation (79). Mechanisms of proteostasis, including autophagy, are attractive drug targets that have the potential to clear multiple pathologies (80).
There are several drugs in clinical testing targeting mechanisms of proteostasis. Nilotinib is an oral Abl tyrosine kinase inhibitor used to treat chronic myeloid leukemia that induces autophagy and leads to the death of rapidly dividing cells (81). Nilotinib is currently in Phase 3 testing for AD (NCT05143528). In addition, rapamycin is a small molecule mTOR inhibitor and trehalose injection is a small molecule approach that activates the transcription factor EB. Both rapamycin and trehalose injections are hypothesized to induce autophagy and are in Phase 2 clinical trials for AD (NCT04629495, NCT05332678). The outcome measures used in these studies predominantly address safety, tolerability, and cognition. Having more specific markers of target engagement to inform exposure and downstream biological effects would greatly strengthen decision making for clinical trials testing therapeutics targeting autophagy.
Biomarkers of autophagy are actively being identified and evaluated. CSF concentrations of p62, a marker of autophagic flux, were increased in AD compared to controls (82). Plasma levels of autophagy-related gene 5 (ATG5) were elevated in patients with dementia or MCI compared with control subjects (83). Beclin1, a key protein involved in autophagy, is altered in traumatic brain injury (TBI) (84), PD (85), and AD (86). Other markers such as LC3B, LAMP-2, and ATG7 are being explored as markers of autophagy (85, 87). While progress is being made to explore novel markers of proteostasis, more research is needed to better understand how these biomarkers relate to Alzheimer’s pathology and progression. Better biomarkers will be important for preclinical and clinical development of specific drugs targeting mechanisms of proteostasis.
Synaptic dysfunction and loss of cortical synapses
AD is marked by progressive neuronal and synaptic loss that strongly correlate with cognitive decline. The presence of pathological forms of Aβ or tau are sufficient to produce synaptic dysfunction and synaptotoxicity, but the precise underlying mechanisms behind synaptic dysfunction are not clear. One hypothesis is that buildup of AD-associated pathology drives changes in microglia which are then prompted to ingest and eliminate synapses (88). Synaptic dysfunction could serve as a marker of age-related cognitive dysfunction, possibly preceding or even progressing independently from AD pathogenesis (89). Synaptic dysfunction and loss serve as novel and promising drug targets that need biomarkers for effective development.
Synaptic health can be evaluated by measuring proteins in the CSF from both pre-synaptic and post-synaptic compartments. Pre-synaptic biomarkers including synaptosomal-associated protein of 25 kDa (SNAP-25), synaptotagmin, growth associated protein 43 (GAP-43), and synaptophysin are all increased in AD. These increased levels are hypothesized to result from synaptic breakdown and clearance through the CSF (88). Neuronal pentraxins are extracellular scaffolding proteins emerging as a potential biomarker of synaptic dysfunction (90). The primary post-synaptic marker is neurogranin, concentrated on dendritic spines in the hippocampus and involved in synaptic plasticity. CSF levels of neurogranin are increased in patients with AD, highly correlated with total tau and p-tau181, and predictive of progression from MCI to AD (91). Interestingly, neurogranin seems to be specific to AD, not other neurodegenerative diseases and correlates with future cognitive decline, glucose metabolism, and brain atrophy at early disease stages (92-94). In normal aging, neurogranin shows a significant relationship with memory performance independent of AD-related biomarkers (89).
Work has also explored whether synaptic markers can be measured in neuronal-derived exosomes. Neuronal-derived exosome levels of synaptophysin, synaptopodin, synaptotagmin-2, neurogranin, GAP-43, and synapsin were lower in AD compared to control participants and decreased years before dementia onset in AD patients (95). These findings suggest that synapses are lost at an accelerated rate in AD. Further development of less invasive measures is needed to support clinical trials of novel therapeutics targeting synaptic function.
In addition to fluid markers, there are now PET tracers available that bind to the synaptic vesicle glycoprotein 2A (SV2A), a transmembrane protein of synaptic vesicles present in synaptic terminals and involved in neurotransmitter release. Levels of SV2A were found to be decreased in postmortem tissue and in signal from SV2A PET scans in AD (96). While promising, further work is needed to validate SV2A PET ligands for use as a biomarker of synaptic health in clinical trials for AD. In 2022, the Foundation for the National Institutes of Health launched a pre-competitive, 3-year initiative that brings together key stakeholders to advance the development of a SV2A PET ligand as a biomarker that could be used to track disease progression and as an outcome measure in clinical trials (97). If successful, this initiative and additional validation studies would support the use of SV2A PET ligands for assessing synaptic integrity in drug development.
Several AD clinical trials targeting synaptic mechanisms are currently underway. CT1812, a small molecule sigma2 antagonist designed to prevent oligomeric Aβ from binding at the synapse, is being investigated in two Phase 2 studies in early and mild to moderate AD (NCT03507790, NCT05531656). SV2A PET and other synaptic markers have been used in CT1812’s development strategy (98). In addition, a pivotal Phase 3 clinical trial in patients with mild to moderate AD is testing a novel device aimed to evoke gamma oscillations hypothesized to improve synaptic connections between neurons, activate microglia, and enhance the removal of pathological proteins in the brain (NCT05637801). To support these and future trials, a broader arsenal of biomarkers for synaptic integrity and health will be valuable.
More research is needed to determine which synaptic markers, or combinations of synaptic markers, are most informative for clinical trials testing interventions targeting synaptic protection. Sensitive markers that provide prognostic information before significant synaptic loss are needed to identify patients at the optimal time for intervention. For drugs targeting other mechanisms within the biology of aging such as inflammation or oxidative stress, the relationship between those mechanisms and synaptic loss needs to be better understood. Synaptic markers should also be explored as potential surrogate markers for cognitive decline that may predict clinical efficacy in larger, later stage trials. Blood levels of synaptic proteins have not yet proven reliable (99, 100), but continued efforts to quantify synaptic health through biomarkers in blood will enable less invasive testing and more frequent sampling for clinical trials in AD.
Mitochondrial oxidative stress & metabolic dysfunction
Mitochondria play an essential role in multiple cellular functions including respiration, energy production, cell cycle regulation, and proteostasis (6). Mitochondrial dysfunction is believed to drive aging and contribute to the onset and progression of AD (6, 101). Due to high metabolic demands and low levels of antioxidative defense mechanisms, the brain is particularly vulnerable to oxidative damage. Mitochondrial and metabolic dysfunctions, as well as inadequate antioxidant scavenging, lead to the accumulation of reactive oxygen species (ROS) that can induce oxidative stress, lipid peroxidation, and DNA damage (102-104). Moreover, impaired mitochondrial efficiency and increased ROS production can disrupt glutamatergic signaling, accelerating cognitive decline (105).
Several biomarkers have shown promise in assessing oxidative stress in AD. For instance, the formation of 8-hydroxy-2’deoxyguanosine (8-OHdG) and 8-hydroxy-2’oxyguanisine (8-OHG) from the reaction of free oxygen radicals with DNA and RNA, respectively, are biomarkers for oxidative stress-related DNA damage and are elevated in individuals with AD (106). In a Phase 2a study investigating edaravone in AD, a free radical scavenger approved for the treatment of amyotrophic lateral sclerosis (ALS), CSF and plasma 8-OHdG and 8-OHG are being used as pharmacodynamic primary outcome measures (NCT05323812).
Furthermore, CSF biomarkers of lipid peroxidation such as isoprostanes and neuroprostanes are associated with AD and hold promise as target engagement markers for novel therapeutics targeting mechanisms of oxidative stress (12, 107, 108). Notably, measurement of CSF F4-isoprostanes, which reflect the degree of docosahexaenoic acid (DHA) lipid peroxidation, may be particularly informative due to the association between DHA loss and neurodegeneration (108). Additionally, ROS can impair cell function by forming adducts on proteins. Thus, lipid peroxidation protein adducts, such as isoketals and neuroketals that occur along the isoprostane and neuroprostane pathways, respectively, serve as functional indicators of lipid peroxidation (109, 110).
The unfolded conformational variant of p53 (U-p53), detected in CSF and plasma, has emerged as a potential prognostic and diagnostic biomarker for AD (111). U-p53 levels gradually increase throughout disease progression due to amyloid exposure and oxidative stress and are linked to neurodegeneration (112, 113). A novel antibody, 2D3A8, to detect the AD-specific U-p53 variant has been developed and shows promising performance in supporting diagnosis and predicting AD outcomes. Plasma U-p53 levels have been shown to distinguish AD from MCI and non-AD and reliably predict the onset of AD up to six years before diagnosis (111).
Fluorodeoxyglucose (FDG)-PET, a specialized PET scan measuring glucose metabolism, is widely used to assess cerebral metabolism in AD, and has demonstrated value as a pharmacodynamic and monitoring biomarker in clinical trials (12). For instance, in a Phase 2 trial of the repurposed therapy riluzole, a glutamate modulator approved for ALS, FDG-PET was used to assess the effects on brain metabolic decline in individuals with mild AD. Riluzole slowed brain metabolic decline compared to placebo. FDG-PET scans also correlated with cognitive decline and predicted disease progression (114). Similarly, FDG-PET was employed in a Phase 2 proof-of-concept study of the repurposed drug rasagiline, indicated in PD, to monitor changes in cerebral metabolism between treatment groups (115).
AD is often referred to as “type 3 diabetes” due to the presence of impaired glucose uptake and insulin resistance in the brain (116). Consequently, many trials of new and repurposed agents are focusing on improving brain glucose utilization and metabolic function (8). While traditional metabolic measures used in diabetes trials primarily target systemic metabolism, the inclusion of FDG-PET has proven valuable in AD studies as a tool to measure brain-specific metabolism. Another biomarker of cellular metabolism in the brain is nicotinamide adenine dinucleotide (NAD+), which can be visualized using magnetic resonance spectroscopy or measured in CSF. NAD+ is a critical factor in mitochondrial and cellular function, and its decline with aging is particularly detrimental to the brain due to the high energy demands of neurons (117-119). Restoring NAD+ levels may have neuroprotective effects by reducing oxidative stress and ameliorating the effects of mitochondrial dysfunction (120). Nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN) are precursors to NAD+ and have been studied as therapeutic strategies for neurodegenerative diseases (121, 122). Currently, a Phase 1/2 study is underway to examine the pharmaceutical-grade NMN known as MIB-626 in Alzheimer’s patients. The primary outcome of this study is changes in CSF levels of MIB-626, while secondary outcomes include changes in CSF levels of NAD+ in the brain and peripheral blood mononuclear cells (NCT05040321).
Furthermore, neural-derived exosomes have been previously used to evaluate the effect of exenatide, a glucagon-like peptide 1 agonist used in type 2 diabetes, on brain insulin signaling pathways in patients with PD. Individuals with PD who received exenatide exhibited greater activation of brain insulin-signaling proteins and downstream effectors compared to their baseline measurements and the placebo group (123). These findings provided valuable mechanistic insights that complemented the clinical outcomes of the trial. There is potential value in harnessing neural- and glial-derived exosomes in AD studies to measure neuropathological changes over time. Continued efforts to develop biomarkers for mitochondria and metabolic dysfunction will be important.
Epigenetics
Emerging evidence suggests that epigenetic factors play a role in the development of AD. Epigenetic changes alter gene expression by making structural and biochemical changes to chromatin or marking histones that package DNA while leaving the DNA sequence unchanged (124, 125). These changes can include DNA methylation, histone modifications (histone methylation, acetylation, glycosylation, ubiquitination, phosphorylation), and non-coding RNA changes.
A prominent biomarker of aging is the Horvath clock, an estimation of biological age based on DNA methylation markers. This epigenetic clock has been linked to the neuropathology present in AD and is related to decline in cognitive and memory function (126). Studies using post-mortem tissue from patients with AD suggest that histone markers are correlated with abnormal tau phosphorylation and Aβ plaques (127) and levels of histone deacetylases (HDACs) are increased in MCI and AD compared to controls (128). However, use of the [11C]Martinostat PET ligand, selective for class I histone deacetylase, showed that HDAC I levels were reduced in patients with AD which mediated the harmful effects of Aβ and tau on brain atrophy and cognitive impairment (129). Emerging approaches are exploring whether epigenetic mechanisms can be measured in blood. These include quantifying patterns of cell-free DNA methylation, fragmentation, and histone markers (130), capturing methylcytosines in mitochondrial DNA using next generation sequencing, and using long noncoding RNA panels (131).
In the current AD clinical trial pipeline, one therapeutic targeting an epigenetic mechanism is lamivudine, a small molecule HIV nucleoside analog reverse transcriptase inhibitor currently in an open-label, one arm Phase 2 study (NCT04552795). The primary outcome is a change in reverse transcriptase activity in blood and CSF to measure target engagement and an assay to quantify lamivudine’s CNS penetrance. Other approaches, such as targeting HDACs or histone acetyltransferases are in earlier stages of development (132, 133).
Further research is needed to determine whether epigenetic changes play a causative role in dementia development or serve as an adaptation to other initiating pathologies. Epigenetic modulation is a promising therapeutic target for AD, but further study is required to mature drugs and biomarkers targeting epigenetic changes in dementia and aging.
Future Directions
The development of novel drugs targeting the biology of aging relies on the integration of a diverse set of biomarkers in clinical trials. Multiple modalities are being pursued, including fluid analyses (CSF, blood), neuroimaging (PET, MRI), retinal scans, analyses of digital signatures to identify reliable biomarkers, and others, each with specific advantages and limitations. Analytes measured in CSF can be specific to the brain and spinal cord but require an invasive lumbar puncture and cannot provide information regarding pathology location. Analytes measured in blood are affordable, accessible, and scalable but suffer from rapid degradation and combination with peripheral components. Neuroimaging provides spatial localization information that CSF and blood markers lack. Structural MRI provides high spatial resolution and can capture multiple biomarkers simultaneously (eg, brain volume, cerebrovascular pathology) but lacks molecular specificity. PET tracers allow measurement of specific analytes such as amyloid, tau, and glucose utilization, but are limited to one pathology, are high cost, and require extensive supportive infrastructure.
Recently, a shift to less invasive modalities has been a significant advancement in the development of treatments for neurodegenerative diseases. For example, tofersen, a therapy for ALS patients with a mutation in the superoxide dismutase 1 gene, was approved by the FDA based on a reduction in plasma neurofilament light, a biomarker of neurodegeneration (134). This marked the first drug approval for ALS based on a biomarker. Further, plasma biomarkers are emerging as valuable, lower-cost tools for the investigation of novel therapies in AD. Assessment of plasma Aβ1–42 to Aβ1–40 ratio by mass spectrometric methods can be a sensitive and specific indicator of amyloid load in the brain as measured by PET (135). Moreover, new technologies for ultrasensitive detection such as Single-Molecule Array (SIMOA) as well as the fully automated Elecsys and Lumipulse platforms can detect plasma biomarkers such as Aβ1–40 and Aβ1–42 as well as p-tau181, p-tau217, and neurofilament light (100, 136-141). Other less invasive approaches such as retinal scans and the assessment of digital signatures are starting to show promise. These multiple modalities can be used strategically to complement one another in clinical trials to best enable decision making and optimize learnings from each study.
A broad range of biomarkers are needed to improve the rigor of clinical trials investigating novel therapies targeting the biology of aging. For instance, data from the Phase 3 clinical study of the anti-amyloid antibody donanemab revealed that including tau PET in addition to amyloid PET in trial inclusion criteria improved the ability to identify participants who would benefit most from treatment, compared to using amyloid PET alone (142). These findings underscore the importance of assessing multiple biomarkers during patient selection, particularly when testing drugs that carry a risk of serious side effects. Further, because pathological changes in AD are thought to begin years before symptoms emerge (143), an early biomarker-based diagnosis for AD that extends the use of AD pathology biomarkers to include mechanisms from the biology of aging is needed. Emerging work suggests that there may be AD subtypes that include predominant hyperplasticity, innate immune activation, or BBB dysfunction (144), and many patients also have pathological brain α-synuclein or TDP-43. This variability could impact disease progression, treatment response, and risk of side effects.
In addition, important decisions to continue drug development are made in early phase clinical trials. Thus, biomarker data from early phase studies are critical to elucidate the potential therapeutic value of novel compounds. A diverse set of biomarkers, particularly those specific to the mechanism of action of investigational therapies, should be incorporated in early phase clinical trials to facilitate a robust assessment of target engagement and treatment effects. Such an approach will enable better-informed decision making early in drug development.
To be included in clinical trials and support clinical practice, biomarkers must show reliable evidence for a specific COU, including risk/susceptibility, diagnostic, monitoring, predictive, prognostic, pharmacodynamic, and safety, as defined by the FDA (9). Before a biomarker can be used for a specific COU in a drug clinical trial, several factors must be understood: abundance in the blood, CSF, or brain, dynamic range, change over time, intra-individual variability, and preanalytical factors (12). To build a more robust arsenal of available biomarkers for the biology of aging, significant investments must be made to identify and test these markers in large and diverse cohorts, and to technically validate them for specific COUs. For example, Amyvid, a widely used radioactive agent for amyloid PET, required three clinical studies to validate its utility as a biomarker (145), and extensive efforts were needed to establish amyloid PET as a predictive biomarker for clinically meaningful effects in response to treatment with anti-amyloid antibodies. These investments proved instrumental in understanding both the successes and failures of these treatments. Moreover, in addition to rigorous clinical study, achieving consensus and providing education on interpreting biomarker results across research and clinical settings and in diverse populations will be necessary for successful integration of new biomarkers. Adequate funding is needed to support these efforts, which will help ensure information gained from biomarkers is accurate and reliable. It is important to acknowledge that, after thorough investigation, many emerging biomarkers may not demonstrate the rigor required for use as endpoints in clinical trials.
Investing in biomarker development is crucial to advancing our understanding of how distinct pathologies of the biology of aging progress and interact with one another. Higher levels of microglia activation show significant associations with lower synaptic protein levels and poor cognitive performance (146), but more work is needed to better understand the complex interactions between astrocytes, microglia, and synaptic health in patients. It has also been shown that functional autophagy is critical for healthy synaptic functions including neurotransmission and synaptic plasticity (147), and that epigenetic changes may lead to abnormal synaptic plasticity (148). The progression of AD is complex and multifactorial. Leveraging existing biomarkers as well as identifying and validating new ones will help parse out this complexity and provide insight into who, how, and when to treat.
Conclusion
The recent success achieved with anti-amyloid antibodies has laid a new foundation for Alzheimer’s prevention and treatment. For the first time, the field has therapeutics that may alter the course of the disease. More work is needed to build on this foundation and expand therapeutic options to target the biology of aging. Within this context, precision medicine emerges as a strategy, where tailored combinations of disease-modifying interventions could be offered to prevent, delay, or treat pathologies contributing to AD.
Biomarkers are pivotal to this goal. These tools are increasingly being used to inform critical decisions in drug development, often complementing or substituting traditional clinical outcomes. For example, the FDA relied on amyloid PET as a surrogate endpoint, defined as a substitute for a clinical endpoint that is expected to predict clinical benefit, to inform the accelerated approval of anti-amyloid antibodies, aducanumab and lecanemab.
As we investigate new therapies targeting the biology of aging, a diverse arsenal of biomarkers will be necessary. A holistic approach to biomarker development must be adopted to comprehensively assess brain pathology. There is a need for additional biomarkers to aid in prognosis determination, support diagnosis, and predict treatment response to novel therapies targeting the biology of aging. Efforts should focus on identifying target engagement biomarkers to accurately assess the viability of drug candidates. Further research and validation of biomarkers will provide a deeper understanding of the complex interplay between pathologies of aging and AD, leading to improved diagnostic tools and better therapeutic interventions.
Conflicts of Interest: MO, NB, LN, and KP declare there are no conflicts of interest. HMF receives royalties from the Icahn School of Medicine at Mount Sinai and, in the past 3 years, has received advisory board fees from Alector, Otsuka Lundbeck, LifeWorx, and The Key. The Alzheimer’s Drug Discovery Foundation has funded or cofunded projects to develop or clinically test the following drugs or biomarkers that are discussed in the manuscript: CT1812, dasatinib + quercetin, rasagiline, riluzole, nilotinib, semaglutide, edaravone, gamma frequency device, water permeability mapping MRI, cell-free DNA markers, SV2A PET, mitochondrial DNA biomarker, and long noncoding RNA biomarker.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. 2023 Alzheimer’s disease facts and figures. Alzheimers Dement. 2023;19(4):1598-695 DOI: 10.1002/alz.13016.
2. Dolgin E. Alzheimer’s disease is getting easier to spot. Nature. 2018;559(7715):S10-S2 DOI: 10.1038/d41586-018-05721-w.
3. Sevigny J, Suhy J, Chiao P, Chen T, Klein G, Purcell D, et al. Amyloid PET Screening for Enrichment of Early-Stage Alzheimer Disease Clinical Trials: Experience in a Phase 1b Clinical Trial. Alzheimer Dis Assoc Disord. 2016;30(1):1-7 DOI: 10.1097/WAD.0000000000000144.
4. Budd Haeberlein S, Aisen PS, Barkhof F, Chalkias S, Chen T, Cohen S, et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. The Journal of Prevention of Alzheimer’s Disease. 2022;9(2):197-210 DOI: 10.14283/jpad.2022.30.
5. van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, et al. Lecanemab in Early Alzheimer’s Disease. N Engl J Med. 2023;388(1):9-21 DOI: 10.1056/NEJMoa2212948.
6. Gonzales MM, Garbarino VR, Pollet E, Palavicini JP, Kellogg DL, Jr., Kraig E, Orr ME. Biological aging processes underlying cognitive decline and neurodegenerative disease. J Clin Invest. 2022;132(10) DOI: 10.1172/jci158453.
7. Hara Y, McKeehan N, Fillit HM. Translating the biology of aging into novel therapeutics for Alzheimer disease. Neurology. 2019;92(2):84-93 DOI: 10.1212/wnl.0000000000006745.
8. Cummings J, Zhou Y, Lee G, Zhong K, Fonseca J, Cheng F. Alzheimer’s disease drug development pipeline: 2023. Alzheimers Dement (N Y). 2023;9(2):e12385 DOI: 10.1002/trc2.12385.
9. Group F-NBW. BEST (Biomarkers, EndpointS, and other Tools) Resource. Silver Spring (MD), Bethesda (MD): Food and Drug Administration (US) National Institutes of Health (US); 2016.
10. Cook D, Brown D, Alexander R, March R, Morgan P, Satterthwaite G, Pangalos MN. Lessons learned from the fate of AstraZeneca’s drug pipeline: a five-dimensional framework. Nat Rev Drug Discov. 2014;13(6):419-31 DOI: 10.1038/nrd4309.
11. Califf RM. Biomarker definitions and their applications. Experimental Biology and Medicine. 2018;243(3):213-21 DOI: 10.1177/1535370217750088.
12. Cummings J, Kinney J. Biomarkers for Alzheimer’s Disease: Context of Use, Qualification, and Roadmap for Clinical Implementation. Medicina (Kaunas). 2022;58(7) DOI: 10.3390/medicina58070952.
13. van der Flier WM, Scheltens P. The ATN Framework—Moving Preclinical Alzheimer Disease to Clinical Relevance. JAMA Neurology. 2022;79(10):968-70 DOI: 10.1001/jamaneurol.2022.2967.
14. Friedman LG, McKeehan N, Hara Y, Cummings JL, Matthews DC, Zhu J, et al. Value-Generating Exploratory Trials in Neurodegenerative Dementias. Neurology. 2021;96(20):944-54 DOI: 10.1212/wnl.0000000000011774.
15. Nordstrom AL, Mansson M, Jovanovic H, Karlsson P, Halldin C, Farde L, et al. PET analysis of the 5-HT2A receptor inverse agonist ACP-103 in human brain. Int J Neuropsychopharmacol. 2008;11(2):163-71 DOI: 10.1017/s1461145707007869.
16. Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69 Suppl 1:S4-9 DOI: 10.1093/gerona/glu057.
17. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388-405 DOI: 10.1016/s1474-4422(15)70016-5.
18. Sarlus H, Heneka MT. Microglia in Alzheimer’s disease. J Clin Invest. 2017;127(9):3240-9 DOI: 10.1172/jci90606.
19. Hampel H, Caraci F, Cuello AC, Caruso G, Nisticò R, Corbo M, et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Frontiers in Immunology. 2020;11 DOI: 10.3389/fimmu.2020.00456.
20. Tang Y, Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol. 2016;53(2):1181-94 DOI: 10.1007/s12035-014-9070-5.
21. Rajendran L, Paolicelli RC. Microglia-Mediated Synapse Loss in Alzheimer’s Disease. J Neurosci. 2018;38(12):2911-9 DOI: 10.1523/jneurosci.1136-17.2017.
22. Tournier BB, Tsartsalis S, Ceyzériat K, Garibotto V, Millet P. In Vivo TSPO Signal and Neuroinflammation in Alzheimer’s Disease. Cells. 2020;9(9) DOI: 10.3390/cells9091941.
23. Zhou R, Ji B, Kong Y, Qin L, Ren W, Guan Y, Ni R. PET Imaging of Neuroinflammation in Alzheimer’s Disease. Frontiers in Immunology. 2021;12 DOI: 10.3389/fimmu.2021.739130.
24. Gratuze M, Leyns CEG, Holtzman DM. New insights into the role of TREM2 in Alzheimer’s disease. Mol Neurodegener. 2018;13(1):66 DOI: 10.1186/s13024-018-0298-9.
25. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 Variants in Alzheimer’s Disease. New England Journal of Medicine. 2012;368(2):117-27 DOI: 10.1056/NEJMoa1211851.
26. Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. New England Journal of Medicine. 2012;368(2):107-16 DOI: 10.1056/NEJMoa1211103.
27. Wang S, Mustafa M, Yuede CM, Salazar SV, Kong P, Long H, et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. Journal of Experimental Medicine. 2020;217(9) DOI: 10.1084/jem.20200785.
28. Filipello F, Goldsbury C, You SF, Locca A, Karch CM, Piccio L. Soluble TREM2: Innocent bystander or active player in neurological diseases? Neurobiol Dis. 2022;165:105630 DOI: 10.1016/j.nbd.2022.105630.
29. Ward M. A Phase 1 Study of AL002 in Healthy Volunteers. Alzheimer’s Association International Conference; July 26-30; Denver, USA 2021.
30. Semple BD, Kossmann T, Morganti-Kossmann MC. Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J Cereb Blood Flow Metab. 2010;30(3):459-73 DOI: 10.1038/jcbfm.2009.240.
31. Singh S, Anshita D, Ravichandiran V. MCP-1: Function, regulation, and involvement in disease. Int Immunopharmacol. 2021;101(Pt B):107598 DOI: 10.1016/j.intimp.2021.107598.
32. McGrowder DA, Miller F, Vaz K, Nwokocha C, Wilson-Clarke C, Anderson-Cross M, et al. Cerebrospinal Fluid Biomarkers of Alzheimer’s Disease: Current Evidence and Future Perspectives. Brain Sci. 2021;11(2) DOI: 10.3390/brainsci11020215.
33. Kim Y, Park J, Choi YK. The Role of Astrocytes in the Central Nervous System Focused on BK Channel and Heme Oxygenase Metabolites: A Review. Antioxidants (Basel). 2019;8(5) DOI: 10.3390/antiox8050121.
34. Ding ZB, Song LJ, Wang Q, Kumar G, Yan YQ, Ma CG. Astrocytes: a double-edged sword in neurodegenerative diseases. Neural Regen Res. 2021;16(9):1702-10 DOI: 10.4103/1673-5374.306064.
35. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481-7 DOI: 10.1038/nature21029.
36. Lazic A, Balint V, Stanisavljevic Ninkovic D, Peric M, Stevanovic M. Reactive and Senescent Astroglial Phenotypes as Hallmarks of Brain Pathologies. Int J Mol Sci. 2022;23(9) DOI: 10.3390/ijms23094995.
37. Kamphuis W, Middeldorp J, Kooijman L, Sluijs JA, Kooi E-J, Moeton M, et al. Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer’s disease. Neurobiology of Aging. 2014;35(3):492-510 DOI: https://doi.org/10.1016/j.neurobiolaging.2013.09.035.
38. Benedet AL, Milà-Alomà M, Vrillon A, Ashton NJ, Pascoal TA, Lussier F, et al. Differences Between Plasma and Cerebrospinal Fluid Glial Fibrillary Acidic Protein Levels Across the Alzheimer Disease Continuum. JAMA Neurology. 2021;78(12):1471-83 DOI: 10.1001/jamaneurol.2021.3671.
39. Cicognola C, Janelidze S, Hertze J, Zetterberg H, Blennow K, Mattsson-Carlgren N, Hansson O. Plasma glial fibrillary acidic protein detects Alzheimer pathology and predicts future conversion to Alzheimer dementia in patients with mild cognitive impairment. Alzheimer’s Research & Therapy. 2021;13(1):68 DOI: 10.1186/s13195-021-00804-9.
40. Verberk IMW, Laarhuis MB, van den Bosch KA, Ebenau JL, van Leeuwenstijn M, Prins ND, et al. Serum markers glial fibrillary acidic protein and neurofilament light for prognosis and monitoring in cognitively normal older people: a prospective memory clinic-based cohort study. Lancet Healthy Longev. 2021;2(2):e87-e95 DOI: 10.1016/s2666-7568(20)30061-1.
41. Pontecorvo MJ, Lu M, Burnham SC, Schade AE, Dage JL, Shcherbinin S, et al. Association of Donanemab Treatment With Exploratory Plasma Biomarkers in Early Symptomatic Alzheimer Disease: A Secondary Analysis of the TRAILBLAZER-ALZ Randomized Clinical Trial. JAMA Neurology. 2022;79(12):1250-9 DOI: 10.1001/jamaneurol.2022.3392.
42. van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, et al. Lecanemab in Early Alzheimer’s Disease. New England Journal of Medicine. 2022;388(1):9-21 DOI: 10.1056/NEJMoa2212948.
43. Craig-Schapiro R, Perrin RJ, Roe CM, Xiong C, Carter D, Cairns NJ, et al. YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol Psychiatry. 2010;68(10):903-12 DOI: 10.1016/j.biopsych.2010.08.025.
44. Mavroudis I, Chowdhury R, Petridis F, Karantali E, Chatzikonstantinou S, Balmus IM, et al. YKL-40 as a Potential Biomarker for the Differential Diagnosis of Alzheimer’s Disease. Medicina (Kaunas). 2021;58(1) DOI: 10.3390/medicina58010060.
45. Lau DC, Dhillon B, Yan H, Szmitko PE, Verma S. Adipokines: molecular links between obesity and atheroslcerosis. Am J Physiol Heart Circ Physiol. 2005;288(5):H2031-41 DOI: 10.1152/ajpheart.01058.2004.
46. Fernandes A, Tábuas-Pereira M, Duro D, Lima M, Gens H, Santiago B, et al. C-reactive protein as a predictor of mild cognitive impairment conversion into Alzheimer’s disease dementia. Experimental Gerontology. 2020;138:111004 DOI: https://doi.org/10.1016/j.exger.2020.111004.
47. Katsiki N, Ferrannini E. Anti-inflammatory properties of antidiabetic drugs: A “promised land” in the COVID-19 era? J Diabetes Complications. 2020;34(12):107723 DOI: 10.1016/j.jdiacomp.2020.107723.
48. Tao Q, Ang TFA, Akhter-Khan SC, Itchapurapu IS, Killiany R, Zhang X, et al. Impact of C-Reactive Protein on Cognition and Alzheimer Disease Biomarkers in Homozygous Apolipoprotein E ε4 Carriers. Neurology. 2021;97(12):e1243-e52 DOI: 10.1212/wnl.0000000000012512.
49. Gonzales MM, Krishnamurthy S, Garbarino V, Daeihagh AS, Gillispie GJ, Deep G, et al. A geroscience motivated approach to treat Alzheimer’s disease: Senolytics move to clinical trials. Mech Ageing Dev. 2021;200:111589 DOI: 10.1016/j.mad.2021.111589.
50. Roy AL, Sierra F, Howcroft K, Singer DS, Sharpless N, Hodes RJ, et al. A Blueprint for Characterizing Senescence. Cell. 2020;183(5):1143-6 DOI: 10.1016/j.cell.2020.10.032.
51. Gonzales MM, Garbarino VR, Marques Zilli E, Petersen RC, Kirkland JL, Tchkonia T, et al. Senolytic Therapy to Modulate the Progression of Alzheimer’s Disease (SToMP-AD): A Pilot Clinical Trial. J Prev Alzheimers Dis. 2022;9(1):22-9 DOI: 10.14283/jpad.2021.62.
52. Kudlova N, De Sanctis JB, Hajduch M. Cellular Senescence: Molecular Targets, Biomarkers, and Senolytic Drugs. Int J Mol Sci. 2022;23(8) DOI: 10.3390/ijms23084168.
53. Wang AS, Dreesen O. Biomarkers of Cellular Senescence and Skin Aging. Front Genet. 2018;9:247 DOI: 10.3389/fgene.2018.00247.
54. Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke. 2011;42(9):2672-713 DOI: 10.1161/STR.0b013e3182299496.
55. Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain. 2013;136(Pt 9):2697-706 DOI: 10.1093/brain/awt188.
56. Custodia A, Aramburu-Núñez M, Rodríguez-Arrizabalaga M, Pías-Peleteiro JM, Vázquez-Vázquez L, Camino-Castiñeiras J, et al. Biomarkers Assessing Endothelial Dysfunction in Alzheimer’s Disease. Cells. 2023;12(6) DOI: 10.3390/cells12060962.
57. Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80(4):844-66 DOI: 10.1016/j.neuron.2013.10.008.
58. Ku MC, Waiczies S, Niendorf T, Pohlmann A. Assessment of Blood Brain Barrier Leakage with Gadolinium-Enhanced MRI. Methods Mol Biol. 2018;1718:395-408 DOI: 10.1007/978-1-4939-7531-0_23.
59. Ford JN, Zhang Q, Sweeney EM, Merkler AE, de Leon MJ, Gupta A, et al. Quantitative Water Permeability Mapping of Blood-Brain-Barrier Dysfunction in Aging. Front Aging Neurosci. 2022;14:867452 DOI: 10.3389/fnagi.2022.867452.
60. Kockelkoren R, De Vis JB, Mali WP, Hendrikse J, de Jong PA, Rozemuller AM, Koek HL. Hippocampal Calcification on Computed Tomography in Relation to Cognitive Decline in Memory Clinic Patients: A Case-Control Study. PLoS One. 2016;11(11):e0167444 DOI: 10.1371/journal.pone.0167444.
61. Janelidze S, Mattsson N, Stomrud E, Lindberg O, Palmqvist S, Zetterberg H, et al. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology. 2018;91(9):e867-e77 DOI: 10.1212/wnl.0000000000006082.
62. Musaeus CS, Gleerup HS, Høgh P, Waldemar G, Hasselbalch SG, Simonsen AH. Cerebrospinal Fluid/Plasma Albumin Ratio as a Biomarker for Blood-Brain Barrier Impairment Across Neurodegenerative Dementias. Journal of Alzheimer’s Disease. 2020;75:429-36 DOI: 10.3233/JAD-200168.
63. Reinhart WH. Fibrinogen–marker or mediator of vascular disease? Vasc Med. 2003;8(3):211-6 DOI: 10.1191/1358863x03vm494ra.
64. Ryu JK, McLarnon JG. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J Cell Mol Med. 2009;13(9a):2911-25 DOI: 10.1111/j.1582-4934.2008.00434.x.
65. van Oijen M, Witteman JC, Hofman A, Koudstaal PJ, Breteler MM. Fibrinogen is associated with an increased risk of Alzheimer disease and vascular dementia. Stroke. 2005;36(12):2637-41 DOI: 10.1161/01.STR.0000189721.31432.26.
66. Viggars AP, Wharton SB, Simpson JE, Matthews FE, Brayne C, Savva GM, et al. Alterations in the blood brain barrier in ageing cerebral cortex in relationship to Alzheimer-type pathology: a study in the MRC-CFAS population neuropathology cohort. Neurosci Lett. 2011;505(1):25-30 DOI: 10.1016/j.neulet.2011.09.049.
67. Weisel JW. Fibrinogen and fibrin. Adv Protein Chem. 2005;70:247-99 DOI: 10.1016/s0065-3233(05)70008-5.
68. Cortes-Canteli M, Mattei L, Richards AT, Norris EH, Strickland S. Fibrin deposited in the Alzheimer’s disease brain promotes neuronal degeneration. Neurobiol Aging. 2015;36(2):608-17 DOI: 10.1016/j.neurobiolaging.2014.10.030.
69. James BD, Wilson RS, Boyle PA, Trojanowski JQ, Bennett DA, Schneider JA. TDP-43 stage, mixed pathologies, and clinical Alzheimer’s-type dementia. Brain. 2016;139(11):2983-93 DOI: 10.1093/brain/aww224.
70. Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol. 2014;127(6):811-24 DOI: 10.1007/s00401-014-1269-z.
71. Hamilton RL. Lewy Bodies in Alzheimer’s Disease: A Neuropathological Review of 145 Cases Using α-Synuclein Immunohistochemistry. Brain Pathology. 2000;10(3):378-84 DOI: https://doi.org/10.1111/j.1750-3639.2000.tb00269.x.
72. Chung EJ, Babulal GM, Monsell SE, Cairns NJ, Roe CM, Morris JC. Clinical Features of Alzheimer Disease With and Without Lewy Bodies. JAMA Neurol. 2015;72(7):789-96 DOI: 10.1001/jamaneurol.2015.0606.
73. Malampati S, Song J-X, Chun-Kit Tong B, Nalluri A, Yang C-B, Wang Z, et al. Targeting Aggrephagy for the Treatment of Alzheimer’s Disease. Cells. 2020;9(2):311 DOI: 10.3390/cells9020311.
74. Cheng X-T, Zhou B, Lin M-Y, Cai Q, Sheng Z-H. Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. Journal of Cell Biology. 2015;209(3):377-86 DOI: 10.1083/jcb.201412046.
75. Maday S, Wallace KE, Holzbaur ELF. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. Journal of Cell Biology. 2012;196(4):407-17 DOI: 10.1083/jcb.201106120.
76. Morimoto RI, Cuervo AM. Proteostasis and the aging proteome in health and disease. J Gerontol A Biol Sci Med Sci. 2014;69 Suppl 1(Suppl 1):S33-8 DOI: 10.1093/gerona/glu049.
77. Zhang Z, Yang X, Song YQ, Tu J. Autophagy in Alzheimer’s disease pathogenesis: Therapeutic potential and future perspectives. Ageing Res Rev. 2021;72:101464 DOI: 10.1016/j.arr.2021.101464.
78. Nilsson P, Saido TC. Dual roles for autophagy: degradation and secretion of Alzheimer’s disease Aβ peptide. Bioessays. 2014;36(6):570-8 DOI: 10.1002/bies.201400002.
79. Caballero B, Wang Y, Diaz A, Tasset I, Juste YR, Stiller B, et al. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell. 2018;17(1) DOI: 10.1111/acel.12692.
80. Friedman LG, Qureshi YH, Yu WH. Promoting autophagic clearance: viable therapeutic targets in Alzheimer’s disease. Neurotherapeutics. 2015;12(1):94-108 DOI: 10.1007/s13311-014-0320-z.
81. Blay JY, von Mehren M. Nilotinib: a novel, selective tyrosine kinase inhibitor. Semin Oncol. 2011;38 Suppl 1(0 1):S3-9 DOI: 10.1053/j.seminoncol.2011.01.016.
82. Rubino E, Boschi S, Roveta F, Marcinno A, Cermelli A, Borghese C, et al. Investigating p62 Concentrations in Cerebrospinal Fluid of Patients with Dementia: A Potential Autophagy Biomarker In Vivo? Brain Sci. 2022;12(10) DOI: 10.3390/brainsci12101414.
83. Cho S-J, Lim HJ, Jo C, Park MH, Han C, Koh YH. Plasma ATG5 is increased in Alzheimer’s disease. Scientific Reports. 2019;9(1):4741 DOI: 10.1038/s41598-019-41347-2.
84. Au AK, Aneja RK, Bayır H, Bell MJ, Janesko-Feldman K, Kochanek PM, Clark RSB. Autophagy Biomarkers Beclin 1 and p62 are Increased in Cerebrospinal Fluid after Traumatic Brain Injury. Neurocritical Care. 2017;26(3):348-55 DOI: 10.1007/s12028-016-0351-x.
85. Youn J, Lee SB, Lee HS, Yang HO, Park J, Kim JS, et al. Cerebrospinal Fluid Levels of Autophagy-related Proteins Represent Potentially Novel Biomarkers of Early-Stage Parkinson’s Disease. Sci Rep. 2018;8(1):16866 DOI: 10.1038/s41598-018-35376-6.
86. Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118(6):2190-9 DOI: 10.1172/jci33585.
87. Krance SH, Wu CY, Chan ACY, Kwong S, Song BX, Xiong LY, et al. Endosomal-Lysosomal and Autophagy Pathway in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J Alzheimers Dis. 2022;88(4):1279-92 DOI: 10.3233/jad-220360.
88. Tzioras M, McGeachan RI, Durrant CS, Spires-Jones TL. Synaptic degeneration in Alzheimer disease. Nat Rev Neurol. 2023;19(1):19-38 DOI: 10.1038/s41582-022-00749-z.
89. Casaletto KB, Elahi FM, Bettcher BM, Neuhaus J, Bendlin BB, Asthana S, et al. Neurogranin, a synaptic protein, is associated with memory independent of Alzheimer biomarkers. Neurology. 2017;89(17):1782-8 DOI: 10.1212/wnl.0000000000004569.
90. Gómez de San José N, Massa F, Halbgebauer S, Oeckl P, Steinacker P, Otto M. Neuronal pentraxins as biomarkers of synaptic activity: from physiological functions to pathological changes in neurodegeneration. Journal of Neural Transmission. 2022;129(2):207-30 DOI: 10.1007/s00702-021-02411-2.
91. Kester MI, Teunissen CE, Crimmins DL, Herries EM, Ladenson JH, Scheltens P, et al. Neurogranin as a Cerebrospinal Fluid Biomarker for Synaptic Loss in Symptomatic Alzheimer Disease. JAMA Neurol. 2015;72(11):1275-80 DOI: 10.1001/jamaneurol.2015.1867.
92. Kvartsberg H, Lashley T, Murray CE, Brinkmalm G, Cullen NC, Höglund K, et al. The intact postsynaptic protein neurogranin is reduced in brain tissue from patients with familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2019;137(1):89-102 DOI: 10.1007/s00401-018-1910-3.
93. Portelius E, Zetterberg H, Skillbäck T, Törnqvist U, Andreasson U, Trojanowski JQ, et al. Cerebrospinal fluid neurogranin: relation to cognition and neurodegeneration in Alzheimer’s disease. Brain. 2015;138(Pt 11):3373-85 DOI: 10.1093/brain/awv267.
94. Tarawneh R, D’Angelo G, Crimmins D, Herries E, Griest T, Fagan AM, et al. Diagnostic and Prognostic Utility of the Synaptic Marker Neurogranin in Alzheimer Disease. JAMA Neurol. 2016;73(5):561-71 DOI: 10.1001/jamaneurol.2016.0086.
95. Goetzl EJ, Kapogiannis D, Schwartz JB, Lobach IV, Goetzl L, Abner EL, et al. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. Faseb j. 2016;30(12):4141-8 DOI: 10.1096/fj.201600816R.
96. Mecca AP, Chen MK, O’Dell RS, Naganawa M, Toyonaga T, Godek TA, et al. In vivo measurement of widespread synaptic loss in Alzheimer’s disease with SV2A PET. Alzheimers Dement. 2020;16(7):974-82 DOI: 10.1002/alz.12097.
97. FNIH Biomarkers Consortium Project Will Characterize a New Imaging Tool Used to Measure Cognitive Decline in Alzheimer’s Disease Patients [press release]. October, 4, 2022.
98. Pandey K. CSF Proteomics Biomarker Analysis from the SPARC Clinical Trial: To Assess the Effect of the Sigma-2 Receptor (S2R) Modulator CT1812 in Alzheimer’s Disease Patients 2022 [Available from: https://cogrx.com/wp-content/uploads/2022/08/AAIC22-SPARC-virtual-handout.pdf].
99. Camporesi E, Nilsson J, Brinkmalm A, Becker B, Ashton NJ, Blennow K, Zetterberg H. Fluid Biomarkers for Synaptic Dysfunction and Loss. Biomark Insights. 2020;15:1177271920950319 DOI: 10.1177/1177271920950319.
100. Palmqvist S, Insel PS, Stomrud E, Janelidze S, Zetterberg H, Brix B, et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer’s disease. EMBO Mol Med. 2019;11(12):e11170 DOI: 10.15252/emmm.201911170.
101. Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses. 2004;63(1):8-20 DOI: 10.1016/j.mehy.2003.12.045.
102. del Río LA, Corpas FJ, Sandalio LM, Palma JM, Gómez M, Barroso JB. Reactive oxygen species, antioxidant systems and nitric oxide in peroxisomes. Journal of Experimental Botany. 2002;53(372):1255-72 DOI: 10.1093/jexbot/53.372.1255.
103. Egler RA, Fernandes E, Rothermund K, Sereika S, de Souza-Pinto N, Jaruga P, et al. Regulation of reactive oxygen species, DNA damage, and c-Myc function by peroxiredoxin 1. Oncogene. 2005;24(54):8038-50 DOI: 10.1038/sj.onc.1208821.
104. Venkateshappa C, Harish G, Mahadevan A, Srinivas Bharath MM, Shankar SK. Elevated Oxidative Stress and Decreased Antioxidant Function in the Human Hippocampus and Frontal Cortex with Increasing Age: Implications for Neurodegeneration in Alzheimer’s Disease. Neurochemical Research. 2012;37(8):1601-14 DOI: 10.1007/s11064-012-0755-8.
105. Esteras N, Kopach O, Maiolino M, Lariccia V, Amoroso S, Qamar S, et al. Mitochondrial ROS control neuronal excitability and cell fate in frontotemporal dementia. Alzheimers Dement. 2022;18(2):318-38 DOI: 10.1002/alz.12394.
106. Nunomura A, Moreira PI, Castellani RJ, Lee HG, Zhu X, Smith MA, Perry G. Oxidative damage to RNA in aging and neurodegenerative disorders. Neurotox Res. 2012;22(3):231-48 DOI: 10.1007/s12640-012-9331-x.
107. Collin F, Cheignon C, Hureau C. Oxidative stress as a biomarker for Alzheimer’s disease. Biomarkers in Medicine. 2018;12(3):201-3 DOI: 10.2217/bmm-2017-0456.
108. Nourooz-Zadeh J, Liu EHC, Yhlen B, Änggåard EE, Halliwell B. F4 – Isoprostanes as Specific Marker of Docosahexaenoic Acid Peroxidation in Alzheimer’s Disease. Journal of Neurochemistry. 1999;72(2):734-40 DOI: https://doi.org/10.1046/j.1471-4159.1999.0720734.x.
109. Davies SS, Bodine C, Matafonova E, Pantazides BG, Bernoud-Hubac N, Harrison FE, et al. Treatment with a γ-ketoaldehyde scavenger prevents working memory deficits in hApoE4 mice. J Alzheimers Dis. 2011;27(1):49-59 DOI: 10.3233/jad-2011-102118.
110. Zagol-Ikapitte I, Masterson TS, Amarnath V, Montine TJ, Andreasson KI, Boutaud O, Oates JA. Prostaglandin H2-derived adducts of proteins correlate with Alzheimer’s disease severity. Journal of Neurochemistry. 2005;94(4):1140-5 DOI: https://doi.org/10.1111/j.1471-4159.2005.03264.x.
111. Piccirella S, Van Neste L, Fowler C, Masters CL, Fripp J, Doecke JD, et al. A Conformational Variant of p53 (U-p53AZ) as Blood-Based Biomarker for the Prediction of the Onset of Symptomatic Alzheimer’s Disease. J Prev Alzheimers Dis. 2022;9(3):469-79 DOI: 10.14283/jpad.2022.52.
112. Cenini G, Sultana R, Memo M, Butterfield DA. Effects of oxidative and nitrosative stress in brain on p53 proapoptotic protein in amnestic mild cognitive impairment and Alzheimer disease. Free Radic Biol Med. 2008;45(1):81-5 DOI: 10.1016/j.freeradbiomed.2008.03.015.
113. Lanni C, Nardinocchi L, Puca R, Stanga S, Uberti D, Memo M, et al. Homeodomain interacting protein kinase 2: a target for Alzheimer’s beta amyloid leading to misfolded p53 and inappropriate cell survival. PLoS One. 2010;5(4):e10171 DOI: 10.1371/journal.pone.0010171.
114. Matthews DC, Mao X, Dowd K, Tsakanikas D, Jiang CS, Meuser C, et al. Riluzole, a glutamate modulator, slows cerebral glucose metabolism decline in patients with Alzheimer’s disease. Brain. 2021;144(12):3742-55 DOI: 10.1093/brain/awab222.
115. Matthews DC, Ritter A, Thomas RG, Andrews RD, Lukic AS, Revta C, et al. Rasagiline effects on glucose metabolism, cognition, and tau in Alzheimer’s dementia. Alzheimers Dement (N Y). 2021;7(1):e12106 DOI: 10.1002/trc2.12106.
116. Nguyen TT, Ta QTH, Nguyen TKO, Nguyen TTD, Giau VV. Type 3 Diabetes and Its Role Implications in Alzheimer’s Disease. Int J Mol Sci. 2020;21(9) DOI: 10.3390/ijms21093165.
117. Braidy N, Liu Y. Can nicotinamide riboside protect against cognitive impairment? Curr Opin Clin Nutr Metab Care. 2020;23(6):413-20 DOI: 10.1097/mco.0000000000000691.
118. Opitz CA, Heiland I. Dynamics of NAD-metabolism: everything but constant. Biochemical Society Transactions. 2015;43(6):1127-32 DOI: 10.1042/bst20150133.
119. Verdin E. NAD⁺ in aging, metabolism, and neurodegeneration. Science. 2015;350(6265):1208-13 DOI: 10.1126/science.aac4854.
120. Campbell JM. Supplementation with NAD(+) and Its Precursors to Prevent Cognitive Decline across Disease Contexts. Nutrients. 2022;14(15) DOI: 10.3390/nu14153231.
121. Fletcher RS, Lavery GG. The emergence of the nicotinamide riboside kinases in the regulation of NAD+ metabolism. J Mol Endocrinol. 2018;61(3):R107-r21 DOI: 10.1530/jme-18-0085.
122. Freeberg KA, Udovich CAC, Martens CR, Seals DR, Craighead DH. Dietary Supplementation With NAD+-Boosting Compounds in Humans: Current Knowledge and Future Directions. The Journals of Gerontology: Series A. 2023 DOI: 10.1093/gerona/glad106.
123. Athauda D, Gulyani S, Karnati HK, Li Y, Tweedie D, Mustapic M, et al. Utility of Neuronal-Derived Exosomes to Examine Molecular Mechanisms That Affect Motor Function in Patients With Parkinson Disease: A Secondary Analysis of the Exenatide-PD Trial. JAMA Neurol. 2019;76(4):420-9 DOI: 10.1001/jamaneurol.2018.4304.
124. Li Y. Modern epigenetics methods in biological research. Methods. 2021;187:104-13 DOI: 10.1016/j.ymeth.2020.06.022.
125. Wang F, Chen D, Wu P, Klein C, Jin C. Formaldehyde, Epigenetics, and Alzheimer’s Disease. Chem Res Toxicol. 2019;32(5):820-30 DOI: 10.1021/acs.chemrestox.9b00090.
126. Levine ME, Lu AT, Bennett DA, Horvath S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging (Albany NY). 2015;7(12):1198-211 DOI: 10.18632/aging.100864.
127. Narayan PJ, Lill C, Faull R, Curtis MA, Dragunow M. Increased acetyl and total histone levels in post-mortem Alzheimer’s disease brain. Neurobiol Dis. 2015;74:281-94 DOI: 10.1016/j.nbd.2014.11.023.
128. Mahady L, Nadeem M, Malek-Ahmadi M, Chen K, Perez SE, Mufson EJ. Frontal Cortex Epigenetic Dysregulation During the Progression of Alzheimer’s Disease. J Alzheimers Dis. 2018;62(1):115-31 DOI: 10.3233/jad-171032.
129. Pascoal TA, Chamoun M, Lax E, Wey H-Y, Shin M, Ng KP, et al. [11C]Martinostat PET analysis reveals reduced HDAC I availability in Alzheimer’s disease. Nature Communications. 2022;13(1):4171 DOI: 10.1038/s41467-022-30653-5.
130. Fox-Fisher I, Shemer R, Dor Y. Epigenetic liquid biopsies: a novel putative biomarker in immunology and inflammation. Trends Immunol. 2023;44(5):356-64 DOI: 10.1016/j.it.2023.03.005.
131. Chen L, Guo X, Li Z, He Y. Relationship between long non-coding RNAs and Alzheimer’s disease: a systematic review. Pathology – Research and Practice. 2019;215(1):12-20 DOI: https://doi.org/10.1016/j.prp.2018.11.012.
132. Bondarev AD, Attwood MM, Jonsson J, Chubarev VN, Tarasov VV, Schiöth HB. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. British Journal of Clinical Pharmacology. 2021;87(12):4577-97 DOI: https://doi.org/10.1111/bcp.14889.
133. Pirooznia S, Elefant F. Targeting specific HATs for neurodegenerative disease treatment: translating basic biology to therapeutic possibilities. Frontiers in Cellular Neuroscience. 2013;7 DOI: 10.3389/fncel.2013.00030.
134. FDA Grants Accelerated Approval for QALSODY™ (tofersen) for SOD1-ALS, a Major Scientific Advancement as the First Treatment to Target a Genetic Cause of ALS [press release]. April 25, 2023.
135. Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Doré V, et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature. 2018;554(7691):249-54 DOI: 10.1038/nature25456.
136. Bayoumy S, Verberk IMW, den Dulk B, Hussainali Z, Zwan M, van der Flier WM, et al. Clinical and analytical comparison of six Simoa assays for plasma P-tau isoforms P-tau181, P-tau217, and P-tau231. Alzheimer’s Research & Therapy. 2021;13(1):198 DOI: 10.1186/s13195-021-00939-9.
137. Lue LF, Guerra A, Walker DG. Amyloid Beta and Tau as Alzheimer’s Disease Blood Biomarkers: Promise From New Technologies. Neurol Ther. 2017;6(Suppl 1):25-36 DOI: 10.1007/s40120-017-0074-8.
138. Tatebe H, Kasai T, Ohmichi T, Kishi Y, Kakeya T, Waragai M, et al. Quantification of plasma phosphorylated tau to use as a biomarker for brain Alzheimer pathology: pilot case-control studies including patients with Alzheimer’s disease and down syndrome. Molecular Neurodegeneration. 2017;12(1):63 DOI: 10.1186/s13024-017-0206-8.
139. Wilson EN, Young CB, Ramos Benitez J, Swarovski MS, Feinstein I, Vandijck M, et al. Performance of a fully-automated Lumipulse plasma phospho-tau181 assay for Alzheimer’s disease. Alzheimer’s Research & Therapy. 2022;14(1):172 DOI: 10.1186/s13195-022-01116-2.
140. Vandijck M, Degrieck R, Denoyette M, Delanote J, De Jonge M, De Decker B, et al. Analytical performance of the Lumipulse® G β-Amyloid 1-40 Plasma and Lumipulse® G β-Amyloid 1-42 Plasma RUO assays. Alzheimer’s & Dementia. 2022;18(S6):e068990 DOI: https://doi.org/10.1002/alz.068990.
141. Mielke MM, Syrjanen JA, Blennow K, Zetterberg H, Vemuri P, Skoog I, et al. Plasma and CSF neurofilament light: Relation to longitudinal neuroimaging and cognitive measures. Neurology. 2019;93(3):e252-e60 DOI: 10.1212/wnl.0000000000007767.
142. Lilly’s Donanemab Significantly Slowed Cognitive and Functional Decline in Phase 3 Study of Early Alzheimer’s Disease [press release]. May 3, 2023.
143. Beason-Held LL, Goh JO, An Y, Kraut MA, O’Brien RJ, Ferrucci L, Resnick SM. Changes in brain function occur years before the onset of cognitive impairment. J Neurosci. 2013;33(46):18008-14 DOI: 10.1523/jneurosci.1402-13.2013.
144. Tijms BM, Gobom J, Reus L, Jansen I, Hong S, Dobricic V, et al. Pathophysiological subtypes of Alzheimer’s disease based on cerebrospinal fluid proteomics. Brain. 2020;143(12):3776-92 DOI: 10.1093/brain/awaa325.
145. Highlights of prescribing information: Amyvid 2012 [cited FDA. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202008s000lbl.pdf].
146. Casaletto KB, Lindbergh CA, VandeBunte A, Neuhaus J, Schneider JA, Buchman AS, et al. Microglial Correlates of Late Life Physical Activity: Relationship with Synaptic and Cognitive Aging in Older Adults. The Journal of Neuroscience. 2022;42(2):288-98 DOI: 10.1523/jneurosci.1483-21.2021.
147. Lieberman OJ, Sulzer D. The Synaptic Autophagy Cycle. Journal of Molecular Biology. 2020;432(8):2589-604 DOI: https://doi.org/10.1016/j.jmb.2019.12.028.
148. Mehler MF, Mattick JS. Noncoding RNAs and RNA editing in brain development, functional diversification, and neurological disease. Physiol Rev. 2007;87(3):799-823 DOI: 10.1152/physrev.00036.2006.
© The Authors 2023