
H.R.C. Shanks1,2,3, K.M. Onuska1,2,3, S.M. Massa4,5, T.W. Schmitz1,2,3, F.M. Longo6
1. Schulich School of Medicine and Dentistry, Western University, London, Canada; 2. Robarts Research Institute, Western University, London, Canada; 3. Western Institute for Neuroscience, Western University, London, Canada; 4. Veteran’s Administration Health Care System, San Francisco, USA; 5. Department of Neurology, University of California San Francisco, USA; 6. Department of Neurology and Neurological Sciences, Stanford University, USA
Corresponding Author: Dr. Frank Longo, Department of Neurology and Neurological Sciences, Stanford University, USA, longo@stanford.edu, (650) 724-3172 (office); Dr. Taylor Schmitz, Schulich School of Medicine and Dentistry, Western University, London, Canada, tschmitz@uwo.ca, (519) 661-2111 x80129 (office)
J Prev Alz Dis 2023;4(10):699-705
Published online October 24, 2023, http://dx.doi.org/10.14283/jpad.2023.110
Abstract
Alzheimer’s disease is a neurodegenerative disorder which contributes to millions of cases of dementia worldwide. The dominant theoretical models of Alzheimer’s disease propose that the brain passively succumbs to disruptions in proteostasis, neuronal dysfunction, inflammatory and other processes, ultimately leading to neurodegeneration and dementia. However, an emerging body of evidence suggests that the adult brain is endowed with endogenous mechanisms of resilience which may enable individuals to remain cognitively intact for years despite underlying pathology. In this brief review, we discuss evidence from basic neuroscience and clinical research which demonstrates the existence of endogenous molecular signaling pathways that can promote resilience to neurodegeneration. The p75 neurotrophin receptor provides one such pathway of resilience due to its role as a fundamental signaling switch which determines neuronal survival or degeneration. We highlight a series of preclinical studies targeting the p75 neurotrophin receptor in mouse models which demonstrate resilience to amyloid. We briefly discuss the design and goals of a recent clinical trial of p75 neurotrophin receptor modulation in patients with mild to moderate Alzheimer’s disease. Unique challenges for developing therapeutics and biomarkers which are optimized for targeting and detecting endogenous mechanisms of resilience are also discussed. Altogether, this review motivates further trial work of therapeutics modulating the p75 neurotrophin receptor and other deep biology targets.
Key words: Alzheimer’s disease, resilience, p75 neurotrophin receptor, neuroprotection.
Endogenous mechanisms of brain resilience: a tractable therapeutic target?
Alzheimer’s disease (AD) is a neurodegenerative condition caused by a complex profile of genetic and lifestyle risk factors (1). The dominant theoretical model of AD proposes that the brain passively succumbs to disruptions in proteostasis, neuronal dysfunction, inflammatory and other processes, ultimately leading to neurodegeneration and dementia (2, 3). However, an emerging body of evidence suggests that the adult brain is endowed with endogenous mechanisms of resilience to AD pathology. Longitudinal research from large consortia studies such as the Alzheimer’s Disease Neuroimaging Initiative (4) indicates that clinical AD is preceded by a decades-long clinically silent period (5). During this time, amyloid beta (Aβ) oligomerization triggers a downstream cascade of pathological events, including tau phosphorylation and aggregation, inflammation, neuronal dysfunction, and grey matter loss in localized brain regions (6). Despite the underlying pathological load, clinically detectable cognitive impairment does not appear until neurodegeneration progresses to widespread brain regions (5). Moreover, a study of cognitively normal older adults demonstrated that only about 5% of individuals who have high baseline brain Aβ positron emission tomography (PET) uptake converted to a diagnosis of mild cognitive impairment (MCI) or AD by the ~2 year follow-up visit (7), indicating that Aβ burden is only weakly predictive of disease conversion. This substantial temporal lag between the initial onset of Aβ pathology and the appearance of AD dementia raises the possibility that the brain is equipped with endogenous resilience pathways which promote neuroprotection and actively counteract degenerative processes.
Here, we discuss resistance and resilience to AD pathology using the terminology presented by Arenaza-Urquijo and Vemuri (8). In this framework, resistance refers to lower than expected levels of Aβ and/or tau pathology in an at-risk individual. Resilience refers to an ability of the brain to cope with a high pathological load of Aβ and/or tau, producing better than expected neuronal integrity, synaptic function and cognitive performance.
Given that resilience to AD involves coping with a complex array of pathological elements such as Aβ, tau hyperphosphorylation, glial activation and synaptic loss, candidate resilience pathways likely involve “deep biology” targets (9) which affect an array of distinct fundamental cellular processes including plasticity, cytoskeletal stability, autophagy and inflammatory responses. Further, the strong correlation between increasing age and Aβ burden, (7, 10) hints that the cellular pathways which could promote resilience in the face of increasing pathological Aβ load may center on those which become dysfunctional with aging. Age is the strongest risk factor for sporadic AD (11), and multiple cell types which are selectively affected in presymptomatic AD, such as basal forebrain cholinergic neurons (12, 13), are known to be vulnerable to aging (14).
The potential impact of resilience is further suggested by observations in longitudinal studies in which a small subgroup of individuals harboring abundant Aβ and tau pathology retain normal cognition (15, 16). Notably, morphological studies in subjects with Aβ and tau pathology have revealed a persistence of normal densities of dendritic spines in those retaining normal cognition compared to the typical loss of spine density found in AD subjects with dementia (17). The conversion from a trajectory of physiologic aging with “normal” Aβ accumulation may therefore stem from a failure of resilience pathways to further counteract an increasing pathogenic load. Thus, if pathways which promote cellular resilience to AD pathology can be identified, this may create new avenues for the development of neuroprotective, disease-modifying AD therapeutics.
In this brief review, we discuss evidence from basic neuroscience and clinical research which demonstrates that molecular signaling pathways can promote resilience to neurodegeneration under high Aβ load. We then focus on the p75 neurotrophin receptor (p75NTR) as one such critical pathway which could be targeted to promote resilience. Lastly, we conclude with a discussion of a recent phase 2a clinical trial in which targeting of p75NTR with LM11A-31 was assessed.
Resilience to autosomal dominant AD
The importance of the brain’s endogenous pathways for resilience in maintaining sustained cognitive function despite underlying AD pathology has been highlighted through a recent case study of autosomal dominant AD (18). An individual remained cognitively intact for approximately 20 years past the expected onset of dementia, due to a gain of function mutation (RELN-COLBOS) in the RELN gene. The RELN gene encodes the “deep biology” protein (9) reelin, which regulates diverse intracellular signaling pathways important in the structural and functional maintenance of neurons (19). Reelin expression in hippocampal subfields appears to decrease with increasing age from early development to adulthood (20), and regulates neuronal migration, cytoskeletal stability, dendritic spines, synaptic plasticity, neurotransmitter release and tau hyperphosphorylation (19, 21, 22). In Aβ mouse model studies, overexpression of RELN confers resilience of dendritic spines to Aβ accumulation (23).
In the context of autosomal dominant AD, the RELN-COLBOS mutation altered the distribution of tau pathology, reducing tangles in the entorhinal cortex and sparing cognitive function. Notably, these effects were produced without affecting overall Aβ load, indicating that the RELN-COLBOS mutation was able to partially uncouple the relationship between Aβ pathology and downstream tau accumulation (18). This effect was associated with a reduction in Aβ-associated loss of glucose metabolism, as measured by 18F-Fluorodeoxyglucose PET, consistent with a sparing of loss of synaptic function. Further investigation indicated that the RELN-COLBOS mutation led to increased availability of the reelin protein to its dual receptor system. This could potentially result in increased downstream signaling with favorable modulation of signaling intermediates such as the apoptosis inhibitor protein kinase B (AKT), the tau kinase Glycogen Synthase Kinase 3 Beta (GSK3β) and the actin-binding protein cofilin. These signaling elements contribute to regulation of tau phosphorylation and synaptic integrity (21).
p75 neurotrophin receptor modulates resilience pathways
The recent discovery of the RELN-COLBOS mutation, along with its known signaling mechanisms, builds on an existing body of work which suggests that if the brain’s endogenous mechanisms of resilience can be promoted – either genetically or pharmacologically – the brain may be able to counteract or cope with an increasing pathological load, promoting better cognitive performance and prolonging functional independence for those with AD.
One focus of this prior research on resilience pathways has been the p75NTR. p75NTR is a member of the tumor necrosis factor family of receptors, and its intracellular signaling network is at the core of several fundamental resilience pathways regulating functions including cell survival, plasticity, and synaptic integrity (24–26). For a more thorough review of p75NTR functions, readers are directed to (27–30). p75NTR can be expressed by neurons, microglia and astrocytes, which we briefly discuss below.
Neuronal p75NTR expression and signaling
p75NTR signaling mediates age-related deficits in neuronal structure (31) and function (26), and its downstream signaling network has substantial overlap with pathways regulated by reelin, including the regulation of AKT, tau kinases and cofilin (19, 32, 33). In the adult brain, p75NTR is expressed by multiple neuronal populations with relatively high levels in basal forebrain cholinergic and hippocampal pyramidal neurons (34–37). Lower but detectable expression in entorhinal layer 2, cortical layer 3 and 5, locus coeruleus and other neurons has also been documented (38, 39). In AD and other pathological states, p75NTR expression is upregulated in neurons and detectable in glial and other cells (37, 40, 41).
p75NTR regulates both trophic and degenerative signaling pathways (Fig. 1), depending on the cell type and context (27, 42). In neurons, p75NTR generally promotes degenerative signaling pathways when unliganded or when interacting with pro-neurotrophins and Sortilin or the related SorCS2 receptors (33). Although p75NTR can have trophic effects when interacting with tropomyosin receptor kinases (Trk) receptors and mature neurotrophins (43), dysfunctional growth factor metabolism in AD results in decreased Trk expression and an increased ratio of pro-neurotrophins to mature neurotrophins (44, 45). This produces a shift in p75NTR signaling, promoting degenerative pathways leading to the collapse of dendritic spines, reduced axonal integrity and apoptosis.
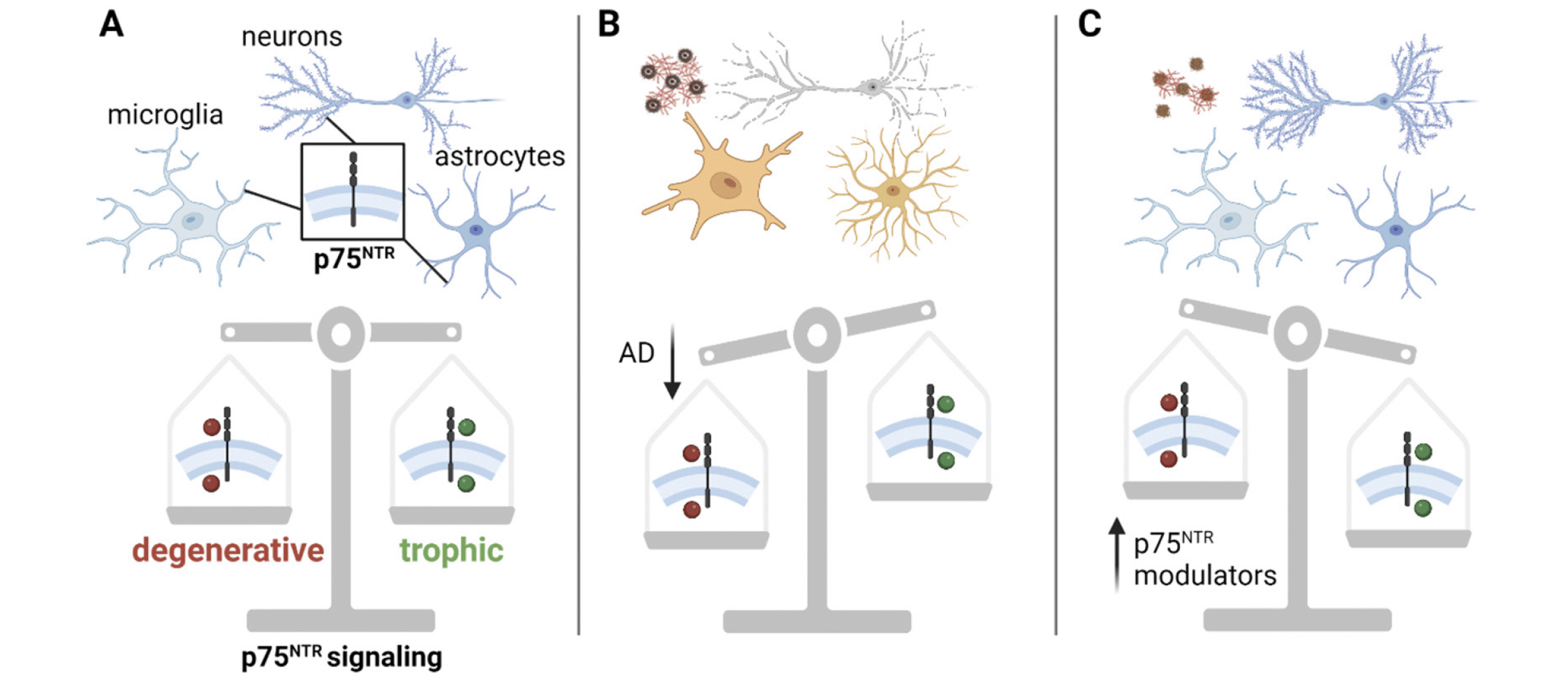
Figure 1. p75NTR regulates cellular fate. A) p75NTR (boxed inset) is expressed in the membrane of neurons, microglia and astrocytes. It acts as a master switch between cell death pathways (red circles) and cell survival pathways (green circles). B) In the AD molecular background (amyloid, tau, microglial and astrocyte reactivity), p75NTR favors degenerative signaling pathways (red circles), yielding neuronal and synaptic degeneration. C) In the AD molecular background, p75NTR modulators bias p75NTR to downregulate degenerative signaling pathways, producing neuronal spine stability, reduced accumulation of pathological tau, and reduced glial inflammatory mechanisms
AD = Alzheimer’s disease; p75NTR = p75 neurotrophin receptor. Created with BioRender.com
Glial p75NTR expression and signaling
p75NTR is expressed on glia during development, where it is involved in myelination, differentiation and cell cycle control (46). Under physiological conditions, glial cells in the adult brain, such as oligodendrocytes, may also express p75NTR to facilitate maintenance of axonal myelination (46). Acquired brain injuries in the adult brain elicit significant upregulation of p75NTR on microglia and astrocytes. In these conditions, increased p75NTR expression and function in glial cells appears to exacerbate pro-inflammatory responses (41, 47, 48). In AD, elevated pro-inflammatory responses due to microglial and astrocyte reactivity to Aβ and tau are now well established as a pivotal event in the transition from age-related Aβ pathology to the progression of widespread neuronal damage (49–52). A causal link of p75NTR signaling to this pro-inflammatory glial response in AD pathology has yet to be established in humans. However, preclinical work in mouse models of Aβ (53, 54) and tau (55) pathology has shown that pharmacological modulation of p75NTR signaling significantly attenuates markers of neuroinflammation.
Overall, degenerative pathways under the control of p75NTR overlap substantially with neuronal and glial pathways which are altered in AD. Modulation of p75NTR may therefore influence multiple pathophysiological mechanisms of AD, including formation of pathological tau, neuronal and synaptic degeneration, and inflammation (55). In the next sections, we review preclinical and clinical work which has leveraged this potential strategy through pharmacological modulation of p75NTR signaling.
Targeting p75NTR-mediated resilience pathways for AD treatment
Pre-clinical studies of p75NTR modulator LM11A-31
Given the overlap in p75NTR-mediated and AD degenerative signaling pathways, we previously defined and developed small molecule compounds which selectively interact with p75NTR to downregulate degenerative and upregulate trophic signaling functions (56). LM11A-31 is a small molecule based on the structure of β hairpin loop 1 of nerve growth factor. In aged wild type mice, LM11A-31 prevented age-related atrophy of basal forebrain cholinergic axons (31). In the PS19 tauopathy mouse model, LM11A-31 prevented tau phosphorylation and cleavage, as well as misfolding and accumulation of a broad range of pathological forms of tau including paired helical filaments and the development of tau seeding activity (55). The extensive effects on the reduction of multiple tau pathologic species and mechanisms, along with the general neuroprotective effects beyond tau models, provides an example of overlap between a ‘neuroprotective’ approach and a ‘tau-based’ approach. This overlap and broad range of effects is perhaps a manifestation of affecting pivotal fundamental biological mechanisms. Moreover, consistent with p75NTR expression on glial cells as well as neurons, LM11A-31 attenuates cortical and hippocampal microglial activation (53, 54, 57). Overall, in pre-clinical studies (Table 1), modulation of p75NTR with LM11A-31 prevents both aging and disease-induced activation of degenerative pathways.

Table 1. Main effects of LM11A-31 in models of aging, Aβ and tau pathology and its relevance to human biomarkers
App S = Tg(APP(Swe)); App L/S = Tg(Thy-1-hAPP(Lond/Swe)); P301S(PS19) = Tg(Prnp-MAPT*P301S)PS19Vle/J; U/A = unavailable.
LM11A-31 phase 2a clinical trial design
Based on the evidence of LM11A-31’s target engagement in preclinical work, we recently conducted a randomized, placebo-controlled, double-blinded phase 2a safety and exploratory endpoint trial evaluating a 26-week treatment course of LM11A-31 in participants with mild to moderate AD (EU Clinical Trials 2015-005263-16; ClinicalTrials.gov NCT03069014). Because p75NTR is a deep biology target, a multi-domain biomarker strategy was employed which provided broad coverage of potential mechanisms of action. The trial included CSF biomarkers of core AD pathologies (Aβ42, Aβ40, phosphorylated tau-181, total tau), neurodegeneration (neurofilament), pre- and post-synaptic damage (synaptosomal-associated protein25; SNAP25, synaptotagmin-1; SYT1, neurogranin; NG), inflammation (Chitinase-3-like protein 1; YKL40, soluble triggering receptor expressed on myeloid cells; sTREM2), and acetylcholinesterase activity. Grey matter volume, as measured by structural magnetic resonance imaging (MRI), was used as a surrogate measure of human neuronal/synaptic degeneration (58). Glucose metabolism, as measured by 18F-Fluorodeoxyglucose PET, served as a surrogate measure of human synaptic function (59). Additionally, multiple cognitive endpoints were collected, such as the Mini Mental State Exam and the Alzheimer’s Disease Assessment Scale–Cognitive Subscale.
The importance of multimodal biomarkers in therapeutics targeting neuroprotection
Although the evidence from the RELN-COLBOS case study support the ability of deep biology targets to promote resilience to AD, the complexity of deep biology receptor-mediated signaling pathways creates unique challenges for trial design and understanding drug mechanisms of action. Unlike therapies targeting individual pathological features of AD, such as Aβ or tau monoclonal antibodies, drugs modulating deep biology or neuroprotective targets do not necessarily impact amyloid or pathological tau levels in isolation. Rather, this class of drugs may facilitate neuronal and synaptic integrity, promoting resilience to AD pathology. To examine deep biology target engagement in humans, clinical trials will likely require novel types of biomarkers which are sensitive and specific to synaptic and neuronal integrity, as opposed to individual markers of Aβ and tau pathology. Thus, biomarker strategies may need to be developed that can better detect the broad array of downstream signaling proteins affected by such therapies. For example, to facilitate biomarker selection in clinical trials, pre-clinical studies may benefit from employing reverse-translational techniques which could mirror outcome measures included in a clinical trial. If successful, these biomarkers could then be assessed together in a co-clinical trial evaluating candidate therapeutics.
Given the challenges of detecting a cognitive effect with the relatively short durations and limited scales of phase 1 and 2 studies, the collection of multi-domain biomarker endpoints is especially important for disease-modifying therapies that target neuronal resilience (reviewed in (60, 61)). A neuroprotective therapeutic that slows or reverses the underlying loss of dendritic spines and synapses, reflected by slowing of biomarker progression, may affect cognition in longer time frames. Thus, the convergence of multiple biomarker endpoints supporting an effect of active treatment in phase 1 and 2 trials (even in the absence of a significant cognitive effect) may provide insight into whether a candidate neuroprotective therapeutic should advance to larger trials. This strategy can be broadly applied to clinical trials of neuroprotective therapeutics in various age-related diseases, such as Parkinson’s disease and Lewy body dementia.
Conclusion
In conclusion, pursuing deep biology targets in pharmaceutical development may allow researchers to harness the endogenous systems that promote neuroprotection and resilience to AD, possibly providing more comprehensive single-therapeutic treatment strategies for AD (9). The evidence from the RELN-COLBOS case demonstrates that endogenous resilience pathways can promote resilience to Aβ and/or tau pathology, prolonging cognitive function (18). We have focused on the concept of resilience in this review given that the majority of clinical trials are conducted at stages where substantial underlying Aβ and tau accumulation has already occurred (i.e., MCI or mild AD). However, if clinical efforts can focus on targeting individuals who have biomarkers consistent with very early Aβ pathology, resistance to tau pathology may be feasible, possibly slowing or even preventing AD dementia.
Competing interests: F.M.L. and S.M.M. are listed as inventors on patents related to LM11A-31 that is assigned to the University of North Carolina, University of California, San Francisco and the Dept of Veterans Affairs. They are also entitled to royalties distributed by UC and the VA per their standard agreements. Dr. Longo is a principal of, and has financial interest in PharmatrophiX, a company focused on the development of small molecule ligands for neurotrophin receptors that has licensed several related patents.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. Frisoni GB, Altomare D, Thal DR, Ribaldi F, Van Der Kant R, Ossenkoppele R, et al. The probabilistic model of Alzheimer disease: the amyloid hypothesis revised. Nature Reviews Neuroscience. 2022;23(1): 53–66. https://doi.org/10.1038/s41583-021-00533-w.
2. Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science (New York, N.Y.). 1992;256(5054): 184–185. https://doi.org/10.1126/science.1566067.
3. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Molecular Medicine. 2016;8(6): 595–608. https://doi.org/10.15252/emmm.201606210.
4. Mueller SG, Weiner MW, Thal LJ, Petersen RC, Jack CR, Jagust W, et al. Ways toward an early diagnosis in Alzheimer’s disease: The Alzheimer’s Disease Neuroimaging Initiative (ADNI). Alzheimer’s & Dementia. 2005;1(1): 55–66. https://doi.org/10.1016/j.jalz.2005.06.003.
5. Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. The Lancet Neurology. 2010;9(1): 119–128. https://doi.org/10.1016/S1474-4422(09)70299-6.
6. Long JM, Holtzman DM. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell. 2019;179(2): 312–339. https://doi.org/10.1016/j.cell.2019.09.001.
7. Villemagne VL, Pike KE, Chételat G, Ellis KA, Mulligan RS, Bourgeat P, et al. Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Annals of Neurology. 2011;69(1): 181–192. https://doi.org/10.1002/ana.22248.
8. Arenaza-Urquijo EM, Vemuri P. Resistance vs resilience to Alzheimer disease. Neurology. 2018;90(15): 695–703. https://doi.org/10.1212/WNL.0000000000005303.
9. Longo FM, Massa SM. Next-generation Alzheimer’s therapeutics: Leveraging deep biology. The Journal of Prevention of Alzheimer’s Disease. 2020; 1–2. https://doi.org/10.14283/jpad.2020.30.
10. Therneau TM, Knopman DS, Lowe VJ, Botha H, Graff-Radford J, Jones DT, et al. Relationships between β-amyloid and tau in an elderly population: An accelerated failure time model. NeuroImage. 2021;242: 118440. https://doi.org/10.1016/j.neuroimage.2021.118440.
11. Riedel BC, Thompson PM, Brinton RD. Age, APOE and sex: Triad of risk of Alzheimer’s disease. The Journal of Steroid Biochemistry and Molecular Biology. 2016;160: 134–147. https://doi.org/10.1016/j.jsbmb.2016.03.012.
12. Fernández-Cabello S, Kronbichler M, Van Dijk KRA, Goodman JA, Spreng RN, Schmitz TW, et al. Basal forebrain volume reliably predicts the cortical spread of Alzheimer’s degeneration. Brain. 2020;143(3): 993–1009. https://doi.org/10.1093/brain/awaa012.
13. Schmitz TW, Nathan Spreng R, The Alzheimer’s Disease Neuroimaging Initiative. Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nature Communications. 2016;7(1): 13249. https://doi.org/10.1038/ncomms13249.
14. Grothe M, Heinsen H, Teipel SJ. Atrophy of the cholinergic Basal forebrain over the adult age range and in early stages of Alzheimer’s disease. Biological Psychiatry. 2012;71(9): 805–813. https://doi.org/10.1016/j.biopsych.2011.06.019.
15. Riley KP, Snowdon DA, Desrosiers MF, Markesbery WR. Early life linguistic ability, late life cognitive function, and neuropathology: findings from the Nun Study. Neurobiology of Aging. 2005;26(3): 341–347. https://doi.org/10.1016/j.neurobiolaging.2004.06.019.
16. Corrada MM, Berlau DJ, Kawas CH. A population-based clinicopathological study in the oldest-old: the 90+ study. Current Alzheimer Research. 2012;9(6): 709–717. https://doi.org/10.2174/156720512801322537.
17. Boros BD, Greathouse KM, Gentry EG, Curtis KA, Birchall EL, Gearing M, et al. Dendritic spines provide cognitive resilience against Alzheimer’s disease. Annals of Neurology. 2017;82(4): 602–614. https://doi.org/10.1002/ana.25049.
18. Lopera F, Marino C, Chandrahas AS, O’Hare M, Villalba-Moreno ND, Aguillon D, et al. Resilience to autosomal dominant Alzheimer’s disease in a Reelin-COLBOS heterozygous man. Nature Medicine. 2023; 1–10. https://doi.org/10.1038/s41591-023-02318-3.
19. Wasser CR, Herz J. Reelin: Neurodevelopmental Architect and Homeostatic Regulator of Excitatory Synapses. The Journal of Biological Chemistry. 2017;292(4): 1330–1338. https://doi.org/10.1074/jbc.R116.766782.
20. Despotovski V, Vivekanandarajah A, Waters KA, Machaalani R. Expression of reelin with age in the human hippocampal formation. Hippocampus. 2021;31(5): 493–502. https://doi.org/10.1002/hipo.23310.
21. Doehner J, Knuesel I. Reelin-mediated Signaling during Normal and Pathological Forms of Aging. Aging and Disease. 2010;1(1): 12–29.
22. Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, et al. Direct Binding of Reelin to VLDL Receptor and ApoE Receptor 2 Induces Tyrosine Phosphorylation of Disabled-1 and Modulates Tau Phosphorylation. Neuron. 1999;24(2): 481–489. https://doi.org/10.1016/S0896-6273(00)80861-2.
23. Pujadas L, Rossi D, Andrés R, Teixeira CM, Serra-Vidal B, Parcerisas A, et al. Reelin delays amyloid-beta fibril formation and rescues cognitive deficits in a model of Alzheimer’s disease. Nature Communications. 2014;5(1): 3443. https://doi.org/10.1038/ncomms4443.
24. Coulson EJ, Reid K, Baca M, Shipham KA, Hulett SM, Kilpatrick TJ, et al. Chopper, a New Death Domain of the p75 Neurotrophin Receptor That Mediates Rapid Neuronal Cell Death*. Journal of Biological Chemistry. 2000;275(39): 30537–30545. https://doi.org/10.1074/jbc.M005214200.
25. Demuth H, Hosseini S, Düsedeau HP, Dunay IR, Korte M, Zagrebelsky M. Deletion of p75NTR rescues the synaptic but not the inflammatory status in the brain of a mouse model for Alzheimer’s disease. Frontiers in Molecular Neuroscience. 2023;16: 1163087. https://doi.org/10.3389/fnmol.2023.1163087.
26. Wang Z, Kennedy BK, Wong LW, Sajikumar S. Aging inverts the effects of p75NTR -modulated mTOR manipulation on hippocampal neuron synaptic plasticity in male mice. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2023;37(8): e23067. https://doi.org/10.1096/fj.202201640RRR.
27. Meeker RB, Williams KS. The p75 neurotrophin receptor: at the crossroad of neural repair and death. Neural Regeneration Research. 2015;10(5): 721–725. https://doi.org/10.4103/1673-5374.156967.
28. Underwood CK, Coulson EJ. The p75 neurotrophin receptor. The International Journal of Biochemistry & Cell Biology. 2008;40(9): 1664–1668. https://doi.org/10.1016/j.biocel.2007.06.010.
29. Barker PA. p75NTR Is Positively Promiscuous: Novel Partners and New Insights. Neuron. 2004;42(4): 529–533. https://doi.org/10.1016/j.neuron.2004.04.001.
30. Chao MV. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nature Reviews Neuroscience. 2003;4(4): 299–309. https://doi.org/10.1038/nrn1078.
31. Xie Y, Meeker RB, Massa SM, Longo FM. Modulation of the p75 neurotrophin receptor suppresses age-related basal forebrain cholinergic neuron degeneration. Scientific Reports. 2019;9(1): 5273. https://doi.org/10.1038/s41598-019-41654-8.
32. Deinhardt K, Kim T, Spellman DS, Mains RE, Eipper BA, Neubert TA, et al. Neuronal growth cone retraction relies on proneurotrophin receptor signaling through Rac. Science Signaling. 2011;4(202): ra82. https://doi.org/10.1126/scisignal.2002060.
33. Mufson EJ, Counts SE, Ginsberg SD, Mahady L, Perez SE, Massa SM, et al. Nerve Growth Factor Pathobiology During the Progression of Alzheimer’s Disease. Frontiers in Neuroscience. 2019;13: 533. https://doi.org/10.3389/fnins.2019.00533.
34. Pioro EP, Cuello AC. Distribution of nerve growth factor receptor-like immunoreactivity in the adult rat central nervous system. Effect of colchicine and correlation with the cholinergic system–I. Forebrain. Neuroscience. 1990;34(1): 57–87. https://doi.org/10.1016/0306-4522(90)90304-m.
35. Woolf NJ, Gould E, Butcher LL. Nerve growth factor receptor is associated with cholinergic neurons of the basal forebrain but not the pontomesencephalon. Neuroscience. 1989;30(1): 143–152. https://doi.org/10.1016/0306-4522(89)90360-6.
36. Chakravarthy B, Ménard M, Ito S, Gaudet C, Dal Prà I, Armato U, et al. Hippocampal membrane-associated p75NTR levels are increased in Alzheimer’s disease. Journal of Alzheimer’s disease: JAD. 2012;30(3): 675–684. https://doi.org/10.3233/JAD-2012-120115.
37. Hu XY, Zhang HY, Qin S, Xu H, Swaab DF, Zhou JN. Increased p75(NTR) expression in hippocampal neurons containing hyperphosphorylated tau in Alzheimer patients. Experimental Neurology. 2002;178(1): 104–111. https://doi.org/10.1006/exnr.2002.8018.
38. Mufson EJ, Brashers-Krug T, Kordower JH. p75 nerve growth factor receptor immunoreactivity in the human brainstem and spinal cord. Brain Research. 1992;589(1): 115–123. https://doi.org/10.1016/0006-8993(92)91169-f.
39. Yamuy J, Sampogna S, Chase MH. Neurotrophin-receptor immunoreactive neurons in mesopontine regions involved in the control of behavioral states. Brain Research. 2000;866(1–2): 1–14. https://doi.org/10.1016/s0006-8993(00)02204-6.
40. Roux PP, Colicos MA, Barker PA, Kennedy TE. p75 neurotrophin receptor expression is induced in apoptotic neurons after seizure. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 1999;19(16): 6887–6896. https://doi.org/10.1523/JNEUROSCI.19-16-06887.1999.
41. Xu Z, Shi WH, Xu LB, Shao MF, Chen ZP, Zhu GC, et al. Resident Microglia Activate before Peripheral Monocyte Infiltration and p75NTR Blockade Reduces Microglial Activation and Early Brain Injury after Subarachnoid Hemorrhage. ACS Chemical Neuroscience. 2019;10(1): 412–423. https://doi.org/10.1021/acschemneuro.8b00298.
42. Meeker RB, Williams K. Dynamic nature of the p75 neurotrophin receptor in response to injury and disease. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology. 2014;9(5): 615–628. https://doi.org/10.1007/s11481-014-9566-9.
43. Conroy JN, Coulson EJ. High-affinity TrkA and p75 neurotrophin receptor complexes: A twisted affair. Journal of Biological Chemistry. 2022;298(3): 101568. https://doi.org/10.1016/j.jbc.2022.101568.
44. Mufson EJ, Lavine N, Jaffar S, Kordower JH, Quirion R, Saragovi HU. Reduction in p140-TrkA Receptor Protein within the Nucleus Basalis and Cortex in Alzheimer’s Disease. Experimental Neurology. 1997;146(1): 91–103. https://doi.org/10.1006/exnr.1997.6504.
45. Pentz R, Iulita MF, Ducatenzeiler A, Bennett DA, Cuello AC. The human brain NGF metabolic pathway is impaired in the pre-clinical and clinical continuum of Alzheimers disease. Molecular Psychiatry. 2021;26(10): 6023–6037. https://doi.org/10.1038/s41380-020-0797-2.
46. Cragnolini AB, Friedman WJ. The function of p75NTR in glia. Trends in Neurosciences. 2008;31(2): 99–104. https://doi.org/10.1016/j.tins.2007.11.005.
47. Choi S, Friedman WJ. Inflammatory cytokines IL-1β and TNF-α regulate p75NTR expression in CNS neurons and astrocytes by distinct cell-type-specific signalling mechanisms. ASN neuro. 2009;1(2): e00010. https://doi.org/10.1042/AN20090009.
48. Zhang D, Zhao S, Zhang Z, Xu D, Lian D, Wu J, et al. Regulation of the p75 neurotrophin receptor attenuates neuroinflammation and stimulates hippocampal neurogenesis in experimental Streptococcus pneumoniae meningitis. Journal of Neuroinflammation. 2021;18(1): 253. https://doi.org/10.1186/s12974-021-02294-w.
49. Herrup K. Reimagining Alzheimer’s Disease—An Age-Based Hypothesis. The Journal of Neuroscience. 2010;30(50): 16755–16762. https://doi.org/10.1523/JNEUROSCI.4521-10.2010.
50. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s & Dementia : Translational Research & Clinical Interventions. 2018;4: 575–590. https://doi.org/10.1016/j.trci.2018.06.014.
51. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638): 481–487. https://doi.org/10.1038/nature21029.
52. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. The Lancet. Neurology. 2015;14(4): 388–405. https://doi.org/10.1016/S1474-4422(15)70016-5.
53. Nguyen TVV, Shen L, Vander Griend L, Quach LN, Belichenko NP, Saw N, et al. Small molecule p75NTR ligands reduce pathological phosphorylation and misfolding of tau, inflammatory changes, cholinergic degeneration, and cognitive deficits in AβPP(L/S) transgenic mice. Journal of Alzheimer’s disease: JAD. 2014;42(2): 459–483. https://doi.org/10.3233/JAD-140036.
54. James ML, Belichenko NP, Shuhendler AJ, Hoehne A, Andrews LE, Condon C, et al. [18F]GE-180 PET Detects Reduced Microglia Activation After LM11A-31 Therapy in a Mouse Model of Alzheimer’s Disease. Theranostics. 2017;7(6): 1422–1436. https://doi.org/10.7150/thno.17666.
55. Yang T, Liu H, Tran KC, Leng A, Massa SM, Longo FM. Small-molecule modulation of the p75 neurotrophin receptor inhibits a wide range of tau molecular pathologies and their sequelae in P301S tauopathy mice. Acta Neuropathologica Communications. 2020;8(1): 156. https://doi.org/10.1186/s40478-020-01034-0.
56. Massa SM, Xie Y, Yang T, Harrington AW, Kim ML, Yoon SO, et al. Small, Nonpeptide p75NTR Ligands Induce Survival Signaling and Inhibit proNGF-Induced Death. Journal of Neuroscience. 2006;26(20): 5288–5300. https://doi.org/10.1523/JNEUROSCI.3547-05.2006.
57. Yang T, Tran KC, Zeng AY, Massa SM, Longo FM. Small molecule modulation of the p75 neurotrophin receptor inhibits multiple amyloid beta-induced tau pathologies. Scientific Reports. 2020;10(1): 20322. https://doi.org/10.1038/s41598-020-77210-y.
58. Young PNE, Estarellas M, Coomans E, Srikrishna M, Beaumont H, Maass A, et al. Imaging biomarkers in neurodegeneration: current and future practices. Alzheimer’s Research & Therapy. 2020;12(1): 49. https://doi.org/10.1186/s13195-020-00612-7.
59. Colom-Cadena M, Spires-Jones T, Zetterberg H, Blennow K, Caggiano A, DeKosky ST, et al. The clinical promise of biomarkers of synapse damage or loss in Alzheimer’s disease. Alzheimer’s Research & Therapy. 2020;12(1): 21. https://doi.org/10.1186/s13195-020-00588-4.
60. Cummings J. The Role of Biomarkers in Alzheimer’s Disease Drug Development. In: Guest PC (ed.) Reviews on Biomarker Studies in Psychiatric and Neurodegenerative Disorders. Cham: Springer International Publishing; 2019. p. 29–61. https://doi.org/10.1007/978-3-030-05542-4_2. [Accessed 29th September 2021].
61. Cummings J. Lessons Learned from Alzheimer Disease: Clinical Trials with Negative Outcomes. Clinical and Translational Science. 2018;11(2): 147–152. https://doi.org/10.1111/cts.12491.
62. Knowles JK, Simmons DA, Nguyen TVV, Vander Griend L, Xie Y, Zhang H, et al. Small molecule p75NTR ligand prevents cognitive deficits and neurite degeneration in an Alzheimer’s mouse model. Neurobiology of Aging. 2013;34(8): 2052–2063. https://doi.org/10.1016/j.neurobiolaging.2013.02.015.
63. Simmons DA, Knowles JK, Belichenko NP, Banerjee G, Finkle C, Massa SM, et al. A Small Molecule p75NTR Ligand, LM11A-31, Reverses Cholinergic Neurite Dystrophy in Alzheimer’s Disease Mouse Models with Mid- to Late-Stage Disease Progression. PLoS ONE. 2014;9(8): e102136. https://doi.org/10.1371/journal.pone.0102136.
© The Authors 2023