
D. Angioni1, O. Hansson2, R.J. Bateman3, C. Rabe4, M. Toloue5, J.B. Braunstein6, S. Agus7, J.R. Sims8, T. Bittner4, M.C. Carrillo9, H. Fillit10, C.L. Masters11, S. Salloway12, P. Aisen13, M. Weiner14, B. Vellas1-15, S. Gauthier16 and the EU/US/CTAD Task force
EU/US CTAD Task force: Susan Abushakra (Framingham); Mohammad Afshar (Paris); John Alam (Boston); Alicia Algeciras-Schimnich (Rochester); Sandrine Andrieu (Toulouse); Clive Ballard (Exeter); Amos Baruch (San Francisco); Richard Batrla (Nutley); Monika Baudler (Basel); Joanne Bell (Nutley); Sasha Bozeat (Basel); Dawn Brooks (Indianapolis); Tricia Brooks (Nutley); Szofia Bullain (Basel); Jan Burmeister (Los Angeles); Min Cho (Nutley); Emily Collins (Indianapolis); Gavin Cook (Nutley); Jeffrey Cummings (Las Vegas); Chris Dague (Malvern); Susan De Santi (Nutley); Rachelle Doody (Basel); Billy Dunn (Beltsville); Michael Egan (North Wales); Sven Eriksson (Stockholm); Rianne Esquivel (Malvern); Tom Fagan (Nutley); Phyllis Ferrell (Indianapolis); Michela Gallagher (Baltimore); Anna-Kaija Grönblad (Stockholm); Avis Hains (South San Francisco); Harald Hampel (Nutley); Nanco Hefting (Valby); Suzanne Hendrix (Salt Lake City); Carole Ho (South San Francisco); Helen Hu (Nutley); Zahinoor Ismail (Calgary); Daryl Jones (Nutley); Gene Kinney (San Francisco); Paul Kinnon (Solana Beach); Ricky Kurzman (Nutley); Lars Lannfelt (Uppsala); John Lawson (Malvern); Nathalie LeBastard (Malvern); Valérie Legrand (Nanterre); Nicole Lewandowski (Nutley); Carine Lim (Singapore); Costantine Lyketsos (Baltimore); Donna Masterman (San Francisco); Ming Lu (Indianapolis); Mark Mintun (Indianapolis); José Luis Molinuevo (Valby); Cecilia Monteiro (South San Francisco); Bradford Navia (Cambridge); Tomas Odergren (Stockholm); Gunilla Osswald (Stockholm); Lewis Penny (Aberdeen); Michael Pontecorvo (Indianapolis); Anton Porsteinsson (Rochester); Rema Raman (San Diego); Gesine Respondek (Basel); Larisa Reyderman (Nutley); Sharon Rogers (Los Angeles); Paul Rosenberg (Baltimore); Sharon Rosenzweig-Lipson (Baltimore); Mark Roskey (Burlington); Rubel Carrie (Cambridge ); Ziad Saad (Titusville); Rachel Schindler (New York); Dennis Selkoe (Boston); Melanie Shulman (Cambridge); Kaycee Sink (South San Francisco); Lisa Sipe (San Diego); Daniel Skovronsky (Indianapolis); Elizabeth Somers (Nutley); Maria Soto (Toulouse); Johannes Streffer (Lausane); Pedro Such (Valby); Joyce Suhy (San Mateo); Jacques Touchon (Montpellier); Manu Vandijck (Malvern); Anne White (Indianapolis); David Wilson (Burlington); Wagner Zago (San Francisco); Jin Zhou (Nutley)
1. Gerontopole of Toulouse, Alzheimer’s Disease Research and Clinical Center, Toulouse University Hospital, Toulouse, France; 2. Memory Clinic, Skåne University Hospital, Malmö, Sweden; 3. Washington University School of Medicine, St. Louis, MO, USA; 4. Genentech, A Member of the Roche Group, Basel, Switzerland;
5. Quanterix, Massachusetts, USA; 6. C(2)N Diagnostics, Saint Louis, MO, USA; 7. Diadem, Boston, Massachusetts, USA; 8. Eli Lilly and Company, Indianapolis, IN, USA; 9. Alzheimer’s Association, Chicago, Illinois, USA; 10. Alzheimer’s Drug Discovery Foundation, New York, NY, USA; 11. The Florey Institute of Neuroscience and Mental Health, Australia; 12. Butler Hospital and Warren Alpert Medical School of Brown University, Providence, Rhode Island, USA; 13. USC Alzheimer’s Therapeutic Research Institute, San Diego, CA, USA; 14. University of California, San Francisco, CA, USA; 15. UNM Division of General Internal Medicine and Geriatric Medicine, Albuquerque NM, USA; 16. Department of Neurology and Neurosurgery, and Department of Psychiatry, McGill Centre for Studies in Aging, McGill University, Montreal, QC, Canada.
Corresponding Author: D. Angioni, Gerontopole of Toulouse, Alzheimer’s Disease Research and Clinical Center, Toulouse University Hospital, Toulouse, France, angioni.da@chu-toulouse.fr
J Prev Alz Dis 2023;3(10):418-425
Published online June 13, 2023, http://dx.doi.org/10.14283/jpad.2023.68
Abstract
In randomized clinical trials (RCTs) for Alzheimer’s Disease (AD), cerebrospinal fluid (CSF) and positron emission tomography (PET) biomarkers are currently used for the detection and monitoring of AD pathological features. The use of less resource-intensive plasma biomarkers could decrease the burden to study volunteers and limit costs and time for study enrollment. Blood-based markers (BBMs) could thus play an important role in improving the design and the conduct of RCTs on AD. It remains to be determined if the data available on BBMs are strong enough to replace CSF and PET biomarkers as entry criteria and monitoring tools in RCTs.
Key words: Alzheimer’s disease, randomized clinical trials, screening, monitoring, amyloid, tau.
Introduction
The field of blood-based markers (BBMs) in Alzheimer’s Disease (AD) is rapidly evolving and demonstrating increasingly promising performance (1, 2). The state of scientific development of BBMs (i.e. amyloid-beta isoforms, phosphorylated tau species, combined markers and algorithms) and the next steps needed before their implementation in clinical practice have been discussed in the virtual meeting of the CTAD Task Force which took place in May 2022 (3). Following the encouraging results of Clarity AD trial (4), we have arrived at a pivotal moment for the development of new therapeutic strategies to prevent and delay AD progression. In this context, BBMs could play a central role in improving the design and the conduct of randomized clinical trials (RCTs) on AD (5). Positron emission tomography (PET) and cerebrospinal fluid (CSF) analysis are now well-accepted strategies for the detection and monitoring of AD pathological features. However, PET is costly (e.g. the average amyloid PET cost in US is between 5000 and 7000 dollars), time-consuming, inconvenient (e.g. logistic requiring scanner and tracer timing), and not easily accessible outside of the major metropolitan areas. In addition, widespread use of CSF biomarkers could be problematic due to the expertise needed to perform a lumbar puncture and the invasive nature of sample procurement. The use of less resource-intensive markers as BBMs, potentially able to identify the presence or the absence of the drug target (i.e. amyloidopathy, tauopathy, neurodegeneration, or other) could reduce the cost and burden for the study volunteers, and promote the speed of enrollment (6). Within this framework, some clinical trials are already using BBMs for pre-screening or screening procedures and to monitor the pharmacological effects of the study drug (3). However, it remains to be determined whether the data on BBMs performance available thus far are robust enough to justify BBMs to replace PET and CSF biomarkers as entry and monitoring criteria in RCTs. In order to address these issues, the International CTAD Task Force met in San Francisco on November 29th 2022. Specifically, the CTAD Task Force assessed:
1. The use of BBMs as entry criteria in recent and ongoing RCTs
2. Issues related to the use of BBMs as entry criteria
3. The use of BBMs as monitoring criteria in clinical trials
4. Issues related to the use of BBMs as monitoring criteria
5. The next steps and research prospects for BBMs in clinical trials
Short presentations reflecting the perspectives of academia, pharma industry and assay industry were followed by general discussions. Key points are summarized in Tables 2 and 4.
The use of BBMs as entry criteria in clinical trials
BBMs can be used in clinical trials as pre-screening tools, to rule out participants that are not likely to have pathological features of AD. The use of BBMs does reduce the number of confirmatory exams such as PET (amyloid and/or tau) or CSF analysis needed to corroborate the presence of the study drug target in a given patient. Several ongoing and past clinical trials used BBMs with this aim as documented in the Table 1.
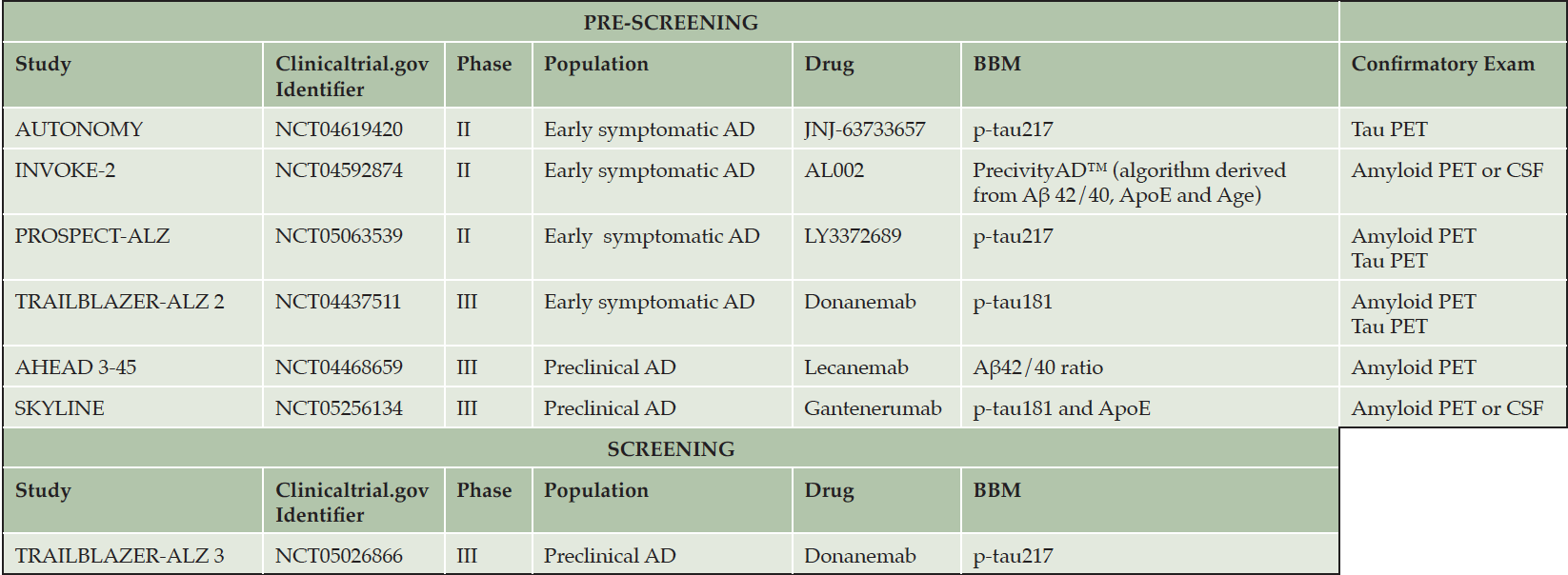
Table 1. Some examples of BBMs use in clinical trials as entry criteria

Table 2. Key points about BBMs use as entry criteria for Alzheimer’s trials
The TRAILBLAZER-ALZ 2 is a phase 3 study designed to evaluate the safety and efficacy of donanemab in early symptomatic AD individuals with confirmed amyloid and intermediate or high tau pathology (7). In this trial, Simoa p-tau181 was explored as a pre-screener. In the early version of the protocol, the positivity of p-tau181 was required before proceeding to amyloid or tau PET. An intermediate analysis has shown that among the 752 patients selected using p-tau181 as a pre-screener, 63 % presented with positivity on both amyloid and tau PET. In contrast, considering 3619 patients screened directly using PET measures, the percentage of individuals with both amyloid and tau PET positivity was markedly lower (37 %) (8). These results indicate that the use of p-tau181 as a pre-screener could reduce screen fails due to amyloid and tau PET negativity and the costs related to PET procedures in clinical trials.
The Janssen Simoa p-tau217 assay is used in the AUTONOMY clinical trial which enrolls individuals with MCI and mild dementia due to AD having a positive tau PET with an intermediate tau burden as a pre-screening tool. Participants with plasma p-tau217 tau levels ≥ of 0.1 pg/ml are allowed to carry on screening procedures with tau PET. An intermediate analysis of 346 patients has shown that 86% of patients with p-tau217 levels above the threshold were tau PET positive and 64% presented an intermediate tau burden (9). Janssen p-tau217 showed excellent performance in predicting amyloid (10, 11, 12) and tau status (13). Janssen p-tau217 might be potentially able to stratify tau pathology and identify early AD participants for anti-tau trials.
Similar results were found in the PROSPECT-ALZ clinical trial, designed to assess the safety and efficacy of the LY3372689, an O-GlcNAcase (OGA) inhibitor, in an early symptomatic AD population. In this trial, considering individuals for which tau PET results are available, 57 % of patients presenting with p-tau217 values above the pre-screening threshold were eligible for randomization following positivity of tau PET (intermediate/high burden) (14).
The plasma amyloid-beta (Aβ) 42/40 ratio, measured using C2N Diagnostics’ liquid chromatography with tandem mass spectrometry (LC-MS/MS) Precivity™ platform, is used to identify participants not likely to have elevated amyloid levels in the AHEAD 3-45 study. The AHEAD 3-45 study consists of two different trials aimed to assess whether lecanemab can prevent cognitive decline and slow tau accumulation in cognitively unimpaired individuals with intermediate (i.e. A3 cohort) and elevated (i.e. A45 cohort) amyloid burden (15). Definitive eligibility for randomization is based on amyloid PET (15).
The INVOKE-2 study is a phase 2 clinical trial designed to assess the efficacy and safety of AL002 in participants with early AD. This study offers an example of the use of a multi-analyte blood test, the PrecivityAD™ test, as a pre-screener (16). PrecivityAD is a validated clinical test that uses C2N’s mass spectrometry methods to generate the Amyloid Probability Score (APS), an algorithm output derived from the combination of the plasma Aβ42/40 ratio, Apolipoprotein E (ApoE) proteotype (i.e. inferred genotype) and the patient’s age (17). The APS reflects the likelihood that an individual being amyloid positive on amyloid PET scan in a scale from 0 to 100. In the INVOKE-2 study amyloid positivity must be confirmed via amyloid PET or CSF analysis.
BBMs can also be used as screening tools in clinical trials for which, in case of the positivity of BBMs, confirmation by PET or CSF analysis is not required. In contrast to their widespread use as pre-screeners, the use of BBMs as screening tools is currently limited to Eli Lilly’s ongoing TRAILBLAZER-ALZ 3 trial, designed to assess the efficacity of donanemab in pre-clinical AD (18).
Issues related to BBMs use as entry criteria
Is concordance with PET and CSF measures high enough?
Currently, CSF markers (e.g. p-tau/Aβ42 or Aβ42/40) and amyloid PET are used interchangeably as entry criteria in AD clinical trials. The neuropathological confirmation by post-mortem examination of Aβ plaques and tau neurofibrillary tangles remains the gold standard for a definitive AD diagnosis (19, 20). While neither CSF analysis or amyloid PET is a true gold standard, they show high concordance with autopsy diagnosis in classifying patients as amyloid positive or negative. This suggests that both CSF and amyloid PET methods effectively capture the underlying amyloid pathology. Multiple BBMs assays have been validated with neuropathological data (21-26). However, other BBMs assays use amyloid PET and CSF as reference standard, thus already limiting their performance metrics due to some errors in PET and CSF assessment. For example, if PET has false negative results, then BBMs may appear artificially higher false positives. For BBMs to replace amyloid PET or CSF, neuropathologic confirmation for these measures is required. In the absence of validation with autopsy data, for BBMs to replace amyloid PET or CSF, a very high predictive value for these measures is needed.
Recent studies suggest that both plasma p-tau markers (specifically p-tau217 and p-tau181 and ratios to their non-phosphorylated forms) and plasma Aβ42/40 assays measured with optimized mass spectrometry-based (MS) methods reach the required high performance in head-to-head studies (10, 12, 27). However, the performance can vary greatly depending on the specific biomarker and assay used with AUCs ranging from 0.65 to 0.96 (10, 12, 27). Combining two BBMs (e.g. Aβ42/40 and p-tau/t-tau) or a single BBM with other clinical factors (e.g. age or ApoE) has been shown to increase the performances (17, 28, 29). To perform well as a screening tool, BBMs should be longitudinally stable at the patient level and over time with limited variance (test-retest variance and variance over time). Plasma Aβ42/40 immunoassays have shown variable results across studies (30). It has been demonstrated that the small difference in immunoassays of Aβ42/40 between PET amyloid positive and negative subjects can result in a lack of robustness (e.g. caused by pre-analytical differences) and reduced clinical performance in less controlled settings (31, 32). The mass spectrometry measurements of Aβ42/40 appear to have greater AUC to predict amyloid PET or a positive CSF profile than immunoassays (30). The analytical stability of these measures appears adequate, with cut point value stability across multiple independent cohort studies (17, 33). More extensive validation of current BBMs will take place in the future and is essential to expand their use in clinical trials. Larger studies are needed to fully characterize BBMs performance and determine for each assay (or combination of assays) whether its concordance with PET and/or CSF performances is high enough to suffice as standalone entry criteria for future clinical trials. However, AUCs approaching 0.95 for several BBM methods versus CSF and amyloid PET (10, 12) suggest feasibility. It is noteworthy that this high level of performance to well-accepted reference standards for amyloid assessment is similar to the performance of CSF relative to PET that was required to achieve recent FDA-clearances of several CSF tests.
Confounding factors and optimal cut points
Currently, cut points are derived from data of previous repositories of plasma collected from patients who have had a PET. It is important to underscore that these cut points were generally established for most assays in research populations (i.e. in most of cases well-educated, White population, with few comorbidities). It remains to be determined whatever various clinical variables (e.g. age, comorbidities, kidney function, drugs, race/ethnicity and cognitive co-pathologies) need to be considered in the interpretation of BBMs levels. Several comorbidities, like ischemic heart disease, stroke, chronic kidney disease, were associated with higher plasma p-tau181 and p-tau217 levels in the Mayo Clinic Study of Aging cohort (34). In the same cohort, chronic kidney disease was also associated with higher levels of plasma Aβ40, Aβ42 and Aβ42/40 ratio measured by Simoa immunoassays (35). In BioFINDER1 and 2 cohorts, creatinine and BMI were associated with Aβ42 and Aβ40 values but not with the Aβ42/40 ratio (36). The mechanism by which comorbidities are associated with changes in cut points is currently unclear. It is unclear if cut points should be adjusted for comorbidities. Furthermore, the prevalence of amyloid positivity has been reported to be lower in Black and other underrepresented populations than in the White population (37). These changes could reflect underlying biology, sampling bias, or other factors. In clinical trials the optimal cut points should account for metrics like positive and negative predictive values (i.e. proportion of patients that are plasma positive/negative and would also be positive/negative with PET). The predictive values depend on the amyloid positivity prevalence in the study population, and reaching high positive predictive values in low prevalence settings requires very high performance of plasma assays. In some settings and using some assays, it has been shown that BBMs may only be useful for ruling out amyloid pathology, while confirmation of amyloid positivity by PET may still be needed in cases tested positive with the plasma biomarker. It is important to note that optimizing for a high positive predictive value (i.e. by using a more extreme cut-off) may exclude a large number of subjects that would have been eligible with amyloid PET, therefore increase the number of patients who need to be screened for a trial, and finally bias the study population (e.g. compared to the population that would have been enrolled with amyloid PET).
The use of BBMs as monitoring criteria in clinical trials
BBMs can be used in clinical trials to assess changes in response to treatment. Due to their well-known properties (i.e. widely accessible, non-invasive, less time-consuming, less expensive), BBMs offer the advantage to be performed more frequently than PET, improving the temporal resolution of monitoring. On the other hand, BBMs do not indicate the neuroanatomic distribution of AD pathology. The main results regarding BBMs use in clinical trials as monitoring criteria are summarized in the Table 3. In the EMERGE and ENGAGE trials, a reduction in plasma p-tau181 levels at week 78 was observed in individuals treated with high doses of aducanumab. Reduced p-tau181 values were correlated with reductions in amyloid PET and with clinical efficacity outcomes (e.g. weak associations with CDR-SB, MMSE, ADAS-COG and ADCS-ADL-MCI scores) in both trials (38). In the BAN2401-G000-201 core trial dose-dependent changes in BBMs (i.e. reduction for p-tau181 and increase Aβ42/40 ratio) were observed in patients treated with lecanemab. These changes were associated with reductions in brain amyloid burden as measured by PET and less cognitive decline over a period of 18 months (39). Interestingly, during the gap period that followed the core phase and preceded the open-label extension (OLE), BBMs tended to return to pre-randomization levels more quickly than amyloid PET: Aβ42/40 decreased by 47%, plasma p-tau181 rose by 24%, while the increase in amyloid burden measured by PET was estimated at 6 centiloids (39). This might suggest that BBMs are more sensitive measures of amyloid re-accumulation compared to amyloid PET. Similar results were found in the phase 3 Clarity AD study (i.e. p-tau181 and glial fibrillary acidic protein reduction and Aβ42/40 increase in the lecanemab arm, with a significant difference with placebo) and the OLE of gantenerumab phase 3 trials Scarlet RoAD and Marguerite RoAD (i.e. reductions in p-tau181 and p-tau217 and increase of Aβ42/40 ratio over 3 years) (4)(40). In a secondary analysis from the TRAILBLAZER-ALZ trial a reduction in plasma p-tau217 and glial fibrillary acidic protein (GFAP) was found in patients treated with donenemab compared to placebo with no significant difference in neurofilament-light chain (NfL) and Aβ42/40 levels between the two arms (41). In this trial, the reduction of p-tau217 and GFAP persisted until the end of the study even in individuals who interrupted the treatment at week 24 following the achievement of targeted amyloid reduction. In TRAILBLAZER-ALZ only changes in NfL showed a significant association with clinical efficacity outcomes (i.e. iADRS). P-tau217 was also used as an exploratory outcome in the TRAILBLAZER-ALZ 4 study which compared donanemab to aducanumab on amyloid plaque clearance in people with early symptomatic AD. Individuals treated with donanemab received 700 mg Q4W for the first 3 doses and the full dose of 1400 mg Q4W thereafter. The full dose of aducanumab (10 mg/kg Q4W) was reached after 24 weeks. In this trial, donanemab, but not aducanumab treatment significantly reduced p-tau217 values at 6 months (42). BBMs are also used as monitoring tools in trials considering other drugs than anti-amyloid antibodies (e.g. small molecules). In this context, in the ALZ-801 phase 2 trial, early AD individuals who are carriers of one or two copies of the ε4 allele of ApoE gene, treated with ALZ-801 presented a reduction in plasma p-tau181 and p-tau181/ Aβ42 levels from baseline to 12 months (43).
Issues related to BBMs use as monitoring criteria
BBMs as monitoring tools at study level
An optimal monitoring BBM to be used in clinical trials should present a large degree of changes during the natural history of AD. Changes in BBMs should be specifically due to AD pathology (and not to other factors). A recent analysis from the BioFINDER cohort showed that plasma p-tau217 had longitudinal increases over a period of 4-6 years in amyloid-positive individuals, with no such changes demonstrated in p-tau231, p-tau181, Aβ42/40, GFAP or NfL that reached a plateau (44). In this study, longitudinal increases of p-tau217 were also associated with declining cognition and brain atrophy in typical AD signature regions. These results were replicated using data from the Wisconsin Registry for Alzheimer’s Prevention.
To be useful as a monitoring tool in RCTs, BBMs values should reflect the drug-induced changes in brain (i.e. reduced AD features as amyloid and tau burden). Recent RCTs (4, 38, 41) have shown that individuals treated with anti-amyloid drugs presented significant differences in BBMs values if compared to placebo (Table 3). However, BBMs levels were not completely “normalized” in the treatment arms to levels of age-matched people without AD. Moreover, it is important to consider that drugs might affect their target differently in the periphery and in the brain. Therapeutic antibodies may alter the half-life of their target in plasma, where large amounts of therapeutic antibodies bind small amounts of amyloid, making the data difficult to interpret. For example, solanezumab was found to inhibit serum-induced degradation of Aβ (45), and anti-tau antibodies to prolong the half-life of tau in blood (46). A potential solution might be to measure a brain-specific protein in blood that is downstream of the drug target instead of the drug target itself (e.g. plasma p-tau in case of anti-amyloid therapy).
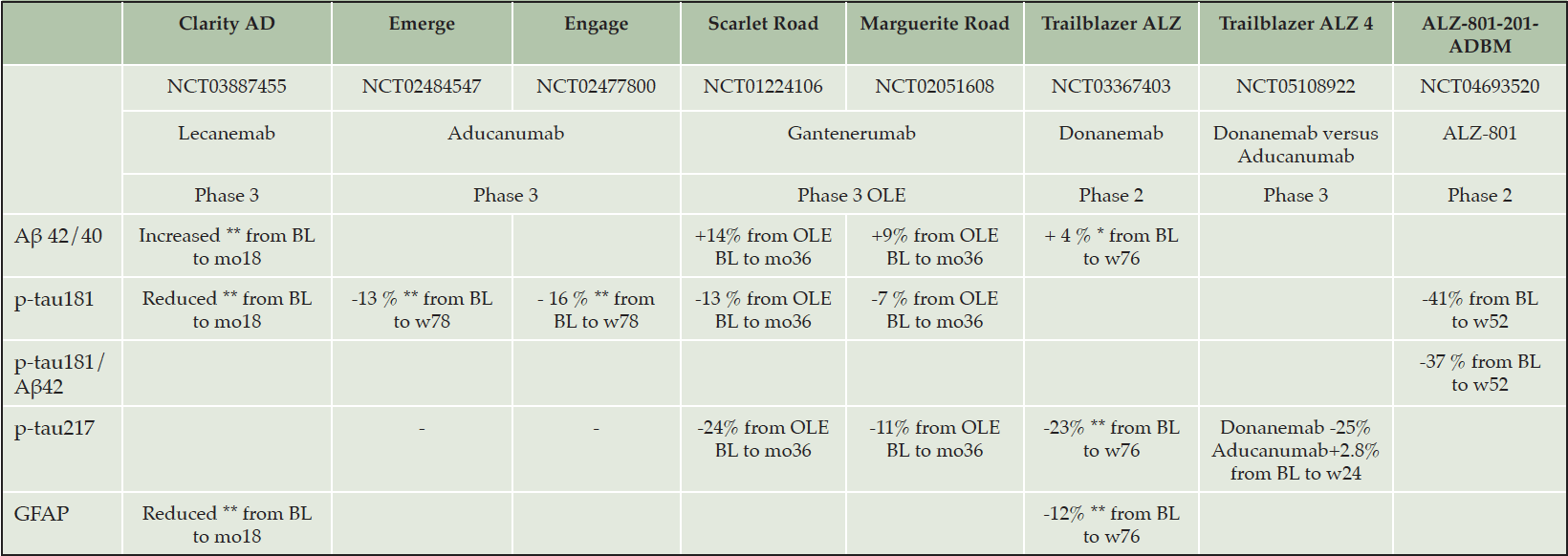
Table 3. Some examples of BBMs use in clinical trials as monitoring criteria
BL, baseline; mo, months; OLE, open label extension; w,week; * no significant difference with placebo arm, ** significant difference with placebo arm

Table 4. Key points about BBMs use to monitor treatment effects in Alzheimer’s trials
Finally, a suitable BBM should not be confounded by other hallmarks of the disease. For example, in case of a monitoring BBM for amyloid, the BBM values should not be impacted by changes in tau pathology. In this case, changes induced by tau pathology but not amyloid pathology could confound the utility of a BBM as a monitoring marker of amyloid. Plasma p-tau species seem to be associated with amyloid and tau pathology (21). This needs to be further investigated.
BBMs as monitoring tools at individual level
BBMs have the potential to provide deterministic qualities at the individual level. If BBMs are validated as monitoring tools at individual level, in AD patients treated with disease-modifying drugs, the monitoring of treatment effects using BBMs may allow dose modification (e.g. less/more frequent or lower/higher dose), discontinuation, switching to other drugs or combinations of drugs. Several BBMs can be considered for this use. Plasma Aβ42/40 has a narrow dynamic range between normal and abnormal, which puts limits on therapeutic response for assays without sufficient precision, particularly at the individual level. NfL is an interesting candidate to monitor the effects of pharmacologic treatments in individuals presenting neurologic diseases involving large myelinated fibers as Multiple Sclerosis and Amyotrophic Lateral Sclerosis (47, 48), but evidence in AD field is lacking at this time. GFAP is a marker of astroglial activation and astrocytosis, non-specific for AD. This could represent a limitation for its use to guide decisions at an individual level. P-tau species look promising but more data are needed to identify and validate for each assay the thresholds corresponding to significant outcomes such as amyloid removal and clinical benefit.
The next steps and research prospects
We are at a turning point in establishing the validity and utility of BBMs in clinical trials as entry criteria. BBMs increase promise and might permit to limit costs, time and complexity of enrollment procedures. However, further research is needed to fully characterize the performance and robustness of BBMs in more diverse populations and settings before they can be considered definitively a substitute for PET imaging or CSF testing as entry criteria. There is an urgent need for standardization of operating procedures to reduce the sources of variability. The question regarding the optimal cut points for each assay is still open and will depend on the specific context of use. More results from samples obtained prospectively are needed to validate, or possibly change, the current cut points. It is extremely desirable to use BBMs to monitor treatment response at a study level, and at a patient level to tailor treatments as is currently done for oncology. BBMs have been mainly used as exploratory outcomes in AD clinical trials so far. More evidence that drug-induced changes in BBMs values are consistently correlated with beneficial changes in clinical outcomes is required to use BBMs as surrogate endpoints in clinical trials. It will be important to understand the extent to which BBM concentrations need to be normalized to be consistently associated with a clinical benefit.
A future goal could be the development of new BBMs having the potential to be used in clinical trials. The AlzoSure® Predict test quantifies the AZ284, a peptide of an unfolded variant of the p53 protein (49). In one study, the AlzoSure test was able to predict cognitive decline to AD-dementia with an AUC > 90 %, regardless of the cognitive status of patients at the time of test. Successful replication in other large cohorts remains to be seen. Combining two or more BBMs may help to increase the accuracy of BBMs. In this context, a new version of the C2N PrecivityAD test (i.e. PrecivityAD2) has been recently developed (50). The PrecivityAD2 combines the measure of the Aβ42/40 ratio and p-tau217 ratio (i.e. tau protein phosphorylated at amino acid 217/ tau protein not phosphorylated at amino acid 217) to obtain a probability score reflecting the likelihood for a patient to be amyloid positive at PET. The PrecivityAD2 showed increased performance (compared to the previous version) with an AUC of 95% and an accuracy of 90% to detect amyloid positivity. Combining a Simoa multiplex of Aβ42/40, GFAP, NfL, together with pTau-181 was recently shown to give superior performance than any of the single biomarkers alone. Independent of age and ApoE status, this multi-marker test achieved an AUC of 91% for detecting amyloid positivity in cognitively normal subjects, and an AUC of 94% for detecting amyloid positivity among MCI patients (51).
BBMs development in RCTs as entry and monitoring criteria may ultimately enable their use in clinical practice when disease-modifying drugs are approved. BBMs have the potential to facilitate the management of disease-modifying drugs in clinical practice for patients, physicians and payers. BBMs will make RCTs more efficient and will facilitate knowledge transfer to clinical practice.
Conflict of interest: The Task Force was partially funded by registration fees from industrial participants. These corporations placed no restrictions on this work. D. Angioni is an investigator in clinical trials sponsored by Roche, Alector, Janssen, Alzheon, Otsuka, Novo Nordisk, UCB Pharma, Medesis Pharma Eisai, Biogen and the CHU of Toulouse. No direct personal benefit is to be declared. O. Hansson has acquired research support (for the institution) from ADx, AVID Radiopharmaceuticals, Biogen, Eli Lilly, Eisai, Fujirebio, GE Healthcare, Pfizer, and Roche. In the past 2 years, he has received consultancy/speaker fees from AC Immune, Amylyx, Alzpath, BioArctic, Biogen, Cerveau, Eisai, Eli Lilly, Fujirebio, Genentech, Merck, Novartis, Novo Nordisk, Roche, Sanofi and Siemens. R. Bateman Washington University and Randall Bateman have equity ownership interest in C2N Diagnostics and Randall Bateman receives income from C2N Diagnostics for serving on the scientific advisory board. Randall Bateman may receive income based on technology licensed by Washington University to C2N Diagnostics. Randall Bateman has received research funding from Avid Radiopharmaceuticals, Janssen, Roche/Genentech, Eli Lilly, Eisai, Biogen, AbbVie, Bristol Myers Squibb and Novartis. Randall Bateman serves on the Roche Gantenerumab Steering Committee as an unpaid member. C. Rabe is a full-time employee of Genentech and owns stock options in F.Hoffmann-LaRoche. M. Toloue is an employee and shareholder in Quanterix Corporation. J.B. Braunstein is a full-time employee of and owns equity in C2N Diagnostics. Sam Agus is an employee of Diadem spA. J.R. Sims is an employee of Eli Lilly and Company: salary and minor stockholder. T. Bittner is a full-time employee of F.Hoffmann-LaRoche and Genentech and owns stock options in F.Hoffmann-LaRoche. M. Carrillo is a full-time employee of the Alzheimer’s Association. She received lead grants (without funding) from NIA LEADS (Co-PI, non salaried). She has served on the scientific advisory board for San Antonio Alz Center EAB, ATRI/ACTC EAB and US POINTER Study DSM. She has a daughter that is a full-time graduate student in the USC Neuroscience program. H. Fillit received royalties from the Icahn School of Medicine at Mount Sinai. He is an unpaid consultant to Biogen. He has received consulting fees from Alector, Otsuka/Lundbeck, and LifeWork. He owns stock options in eFamilyCare. C.L. Master has nothing to declare. S. Salloway was the co-chair of the Investigator Steering Committee for the Aducanumab phase 3 program and he served as a site PI for the aducanumab and lecanemab phase 3 studies, the donanemab phase 2 trial and he was the Project Arm Leader for gantenerumab in DIAN-TU. He has received consulting income from Biogen, Lilly, Roche, Genentech, Bolden, Amylyx, Prothena and Eisai. He has no stock or royalties related to any medication in development. S. Salloway serves on the planning committee for the National Disease Modifying Treatment and Diagnostic Registry Work Group and he is a member of the ADRD Therapeutics Work Group. He is the first author for the report of ARIA in aducanumab phase 3 (Salloway, JAMA Neurology, 2022), the report of gantenerumab and solanezumab in DIAN-TU (Salloway, Nature Medicine, 2021). He is a co-author on the report of the donanemab phase 2 trial (Mintun, NEJM, 2021) and the Aducanumab Appropriate Use Recommendations (Cummings, Journal of the Prevention of Alzheimer’s Disease, 2021). P. Aisen reports collaborations on the development of blood-based markers with C2N, Janssen and Roche, during the conduct of the study; grants from Lilly, Eisai, NIH, Alzheimer’s Association; consultant fees from Merck, Biogen, Abbvie, Genentech, ImmunoBrain Checkpoint, Arrowhead, outside the submitted work. M. Weiner reports, outside the submitted work, grants from National Institutes of Health (NIH)/NINDS/National Institute on Aging (NIA), Department of Defense (DOD), California Department of Public Health (CDPH), University of Michigan, Siemens, Biogen, Hillblom Foundation, Alzheimer’s Association, Johnson & Johnson, Kevin and Connie Shanahan, GE, VUmc, Australian Catholic University, The Stroke Foundation, Veterans Administration; personal fees from Boxer Capital, Cerecin, Clario, Dementia Society of Japan, Eisai, Guidepoint, Health and Wellness Partners, Indiana University, LCN Consulting, Merck Sharp & Dohme Corp., NC Registry for Brain Health, Prova Education, T3D Therapeutics, University of Southern California (USC), WebMD, from China Association for Alzheimer’s Disease (CAAD) , Taipei Medical University, AD/PD Congress, Cleveland Clinic, CTAD Congress, Foundation of Learning, Health Society (Japan), INSPIRE Project; U. Toulouse, Japan Society for Dementia Research, Korean Dementia Society, National Center for Geriatrics and Gerontology (NCGG; Japan), University of Southern California (USC); owns stock-options in Alzeca, Alzheon, ALZ Path, Anven. B Vellas is an investigator in clinical trials sponsored by Biogen, Lilly, Roche, Eisai, Pfizer, Pierre Fabre Pharmaceuticals and the Toulouse University Hospital. He has served as SAB member for Biogen, Alzheon, Green Valley, Norvo Nordisk, Longeveron, Rejuvenate Biomed Clinical Pfizer, Eisai France, Advisory Board Meeting – but received no personal compensation. He has served as consultant and/or SAB member for Roche, Lilly, Eisai, TauX, Cerecin with personal compensation. S. Gauthier is a member of scientific advisory boards of Alzheon, AmyriAD, Biogen Canada, Eisai Canada, Enigma, Lily, Medesis, Roche Canada.
References
1. Teunissen CE, Verberk IMW, Thijssen EH, et al. Blood-based biomarkers for Alzheimer’s disease: towards clinical implementation. Lancet Neurol. 2022;21(1):66-77. doi:10.1016/S1474-4422(21)00361-6
2. Hansson, O. Biomarkers for neurodegenerative diseases. Nat Med 27, 954–963 (2021). https://doi.org/10.1038/s41591-021-01382-x
3. Angioni D, Delrieu J, Hansson O, et al. Blood Biomarkers from Research Use to Clinical Practice: What Must Be Done? A Report from the EU/US CTAD Task Force. J Prev Alzheimers Dis. 2022;9(4):569-579. doi:10.14283/jpad.2022.85
4. Van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M et al. Lecanemab in early Alzheimer’s disease. NEJM 2022; published on line Nov 29th doi 10.1056/NEJMoa2212948.
5. Hansson O, Edelmayer RM, Boxer AL, et al. The Alzheimer’s Association appropriate use recommendations for blood biomarkers in Alzheimer’s disease. Alzheimers Dement. 2022;18(12):2669-2686. doi:10.1002/alz.12756
6. Schindler SE, Li Y, Li M, et al. Using Alzheimer’s disease blood tests to accelerate clinical trial enrollment [published online ahead of print, 2022 Aug 7]. Alzheimers Dement. 2022;10.1002/alz.12754. doi:10.1002/alz.12754
7. Paul Solomon, et al. Symposium 2 Presentation 2; Abstract: Symposia, Conferences, Oral communications: 14th Clinical Trials on Alzheimer’s Disease (CTAD) November 9-12, 2021. J Prev Alzheimers Dis. 2021;8(S1):S2-S72. doi:10.14283/jpad.2021.57
8. TRAILBLAZER-ALZ 2 data, presented at 14th Clinical Trials on Alzheimer’s Disease (CTAD) November 9-12, 2021.
9. Triana-Baltze et al. Oral communication 4 ; Abstract: 15th Conference Clinical Trials Alzheimer’s Disease, November 29- December 2, 2022, San Francisco , USA: Symposia – Oral Communications – Late Breaking News. J Prev Alzheimers Dis. 2022;9(S1):S8-S50. doi: 10.14283/jpad.2022.96. PMID: 36471007; PMCID: PMC9734311.
10. Janelidze S, Bali D, Ashton NJ, Barthélemy NR, Vanbrabant J, Stoops E, Vanmechelen E, He Y, Dolado AO, Triana-Baltzer G, Pontecorvo MJ, Zetterberg H, Kolb H, Vandijck M, Blennow K, Bateman RJ, Hansson O. Head-to-head comparison of 10 plasma phospho-tau assays in prodromal Alzheimer’s disease. Brain. 2022 Sep 10:awac333. doi: 10.1093/brain/awac333. Epub ahead of print. PMID: 36087307
11. Groot C, Cicognola C, Bali D, Triana-Baltzer G, Dage JL, Pontecorvo MJ, Kolb HC, Ossenkoppele R, Janelidze S, Hansson O. Diagnostic and prognostic performance to detect Alzheimer’s disease and clinical progression of a novel assay for plasma p-tau217. Alzheimers Res Ther. 2022 May 14;14(1):67. doi: 10.1186/s13195-022-01005-8. Erratum in: Alzheimers Res Ther. 2022 Jun 13;14(1):82. PMID: 35568889; PMCID: PMC9107269.
12. Ashton NJ, Puig-Pijoan A, Milà-Alomà M, et al. Plasma and CSF biomarkers in a memory clinic: Head-to-head comparison of phosphorylated tau immunoassays [published online ahead of print, 2022 Nov 12]. Alzheimers Dement. 2022;10.1002/alz.12841. doi:10.1002/alz.12841
13. Therriault J, Vermeiren M, Servaes S, et al. Association of Phosphorylated Tau Biomarkers With Amyloid Positron Emission Tomography vs Tau Positron Emission Tomography. JAMA Neurol. 2023;80(2):188-199. doi:10.1001/jamaneurol.2022.4485
14. M. Mintun. Can we use blood biomarkers instead of PET as entry criteria for Alzheimer’s Trials: Point of View from Pharma industry. CTAD Task force. November 29, 2022.
15. Rafii MS, Sperling RA, Donohue MC, et al. The AHEAD 3-45 Study: Design of a prevention trial for Alzheimer’s disease [published online ahead of print, 2022 Aug 15]. Alzheimers Dement. 2022;10.1002/alz.12748. doi:10.1002/alz.12748
16. INVOKE-2 data presented at the Alzheimer’s Association International Conference (AAIC), July 26-30 2021.
17. Hu Y, Kirmess KM, Meyer MR, Rabinovici GD, Gatsonis C, Siegel BA, et al. Assessment of a Plasma Amyloid Probability Score to Estimate Amyloid Positron Emission Tomography Findings Among Adults With Cognitive Impairment. JAMA Netw Open. 2022. Apr 1;5(4):e228392.
18. Tariot et Symposium 2 Presentation 3; Abstract: Symposia, Conferences, Oral communications: 14th Clinical Trials on Alzheimer’s Disease (CTAD) November 9-12, 2021. J Prev Alzheimers Dis. 2021;8(S1):S2-S72. doi:10.14283/jpad.2021.57
19. Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7.
20. Jack CR, Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–216. doi: 10.1016/S1474-4422(12)70291-0
21. Salvadó G, Ossenkoppele R, Ashton NJ, et al. Specific associations between plasma biomarkers and postmortem amyloid plaque and tau tangle loads [published online ahead of print, 2023 Mar 13]. EMBO Mol Med. 2023;e2463. doi:10.15252/emmm.202217123
22. Palmqvist S, Janelidze S, Quiroz YT, et al. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA. 2020;324(8):772-781. doi:10.1001/jama.2020.12134
23. Janelidze S, Mattsson N, Palmqvist S, et al. Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med. 2020;26(3):379-386. doi:10.1038/s41591-020-0755-1
24. Thijssen EH, La Joie R, Wolf A, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat Med. 2020;26(3):387-397. doi:10.1038/s41591-020-0762-2
25. Grothe MJ, Moscoso A, Ashton NJ, et al. Associations of Fully Automated CSF and Novel Plasma Biomarkers With Alzheimer Disease Neuropathology at Autopsy [published online ahead of print, 2021 Jul 15]. Neurology. 2021;97(12):e1229-e1242. doi:10.1212/WNL.0000000000012513
26. Lantero Rodriguez J, Karikari TK, Suárez-Calvet M, et al. Plasma p-tau181 accurately predicts Alzheimer’s disease pathology at least 8 years prior to post-mortem and improves the clinical characterisation of cognitive decline. Acta Neuropathol. 2020;140(3):267-278. doi:10.1007/s00401-020-02195-x
27. Janelidze S, Teunissen CE, Zetterberg H, et al. Head-to-Head Comparison of 8 Plasma Amyloid-β 42/40 Assays in Alzheimer Disease. JAMA Neurol. 2021;78(11):1375–1382. doi:10.1001/jamaneurol.2021.3180
28. Li, Yan, Suzanne E. Schindler, James G. Bollinger, Vitaliy Ovod, Kwasi G. Mawuenyega, Michael W. Weiner, Leslie M. Shaw, et al. “Validation of Plasma Amyloid-β 42/40 for Detecting Alzheimer Disease Amyloid Plaques.” Neurology 98, no. 7 (February 15, 2022): e688–99. https://doi.org/10.1212/WNL.0000000000013211.
29. Cullen NC, Janelidze S, Mattsson-Carlgren N, Palmqvist S, Bittner T, Suridjan I, et al. Test-retest variability of plasma biomarkers in Alzheimer’s disease and its effects on clinical prediction models. Alzheimers Dement. 2022. Jun 14;
30. Brand, Abby L., Paige E. Lawler, James G. Bollinger, Yan Li, Suzanne E. Schindler, Melody Li, Samir Lopez, et al. “The Performance of Plasma Amyloid Beta Measurements in Identifying Amyloid Plaques in Alzheimer’s Disease: A Literature Review.” Alzheimer’s Research & Therapy 14, no. 1 (December 27, 2022): 195. https://doi.org/10.1186/s13195-022-01117-1.
31. Rabe, C, Bittner, T, Jethwa, A, et al. Clinical performance and robustness evaluation of plasma amyloid-β42/40 prescreening. Alzheimer’s Dement. 2022; 1- 10. https://doi.org/10.1002/alz.12801
32. Małgorzata Rózga, Tobias Bittner, Richard Batrla, Johann Karl, Preanalytical sample handling recommendations for Alzheimer’s disease plasma biomarkers, Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring, Volume 11, 2019, Pages 291-300, ISSN 2352-8729, https://doi.org/10.1016/j.dadm.2019.02.002.
33. Fogelman I, West T, Braunstein JB, et al. Independent study demonstrates amyloid probability score accurately indicates amyloid pathology [published online ahead of print, 2023 Mar 28]. Ann Clin Transl Neurol. 2023;10.1002/acn3.51763. doi:10.1002/acn3.51763
34. Mielke MM, Dage JL, Frank RD, et al. Performance of plasma phosphorylated tau 181 and 217 in the community [published correction appears in Nat Med. 2022 Oct 10;:]. Nat Med. 2022;28(7):1398-1405. doi:10.1038/s41591-022-01822-2
35. Syrjanen JA, Campbell MR, Algeciras-Schimnich A, et al. Associations of amyloid and neurodegeneration plasma biomarkers with comorbidities. Alzheimers Dement. 2022;18(6):1128-1140. doi:10.1002/alz.12466
36. Pichet Binette A, Janelidze S, Cullen N, et al. Confounding factors of Alzheimer’s disease plasma biomarkers and their impact on clinical performance [published online ahead of print, 2022 Sep 24]. Alzheimers Dement. 2022;10.1002/alz.12787. doi:10.1002/alz.12787
37. Wilkins CH, Windon CC, Dilworth-Anderson P, et al. Racial and Ethnic Differences in Amyloid PET Positivity in Individuals With Mild Cognitive Impairment or Dementia: A Secondary Analysis of the Imaging Dementia–Evidence for Amyloid Scanning (IDEAS) Cohort Study. JAMA Neurol. 2022;79(11):1139–1147. doi:10.1001/jamaneurol.2022.3157
38. Budd Haeberlein S, Aisen PS, Barkhof F, Chalkias S, Chen T, Cohen S, et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J Prev Alzheimers Dis. 2022;9(2):197–210.
39. McDade E, Cummings JL, Dhadda S, et al. Lecanemab in patients with early Alzheimer’s disease: detailed results on biomarker, cognitive, and clinical effects from the randomized and open-label extension of the phase 2 proof-of-concept study. Alzheimers Res Ther. 2022;14(1):191. Published 2022 Dec 21. doi:10.1186/s13195-022-01124-2
40. Bittner et al. Poster presented at the Alzheimer’s Association International Conference (AAIC) San Diego, July 31 – August 4 2022.
41. Pontecorvo MJ, Lu M, Burnham SC, et al. Association of Donanemab Treatment With Exploratory Plasma Biomarkers in Early Symptomatic Alzheimer Disease: A Secondary Analysis of the TRAILBLAZER-ALZ Randomized Clinical Trial. JAMA Neurol. 2022;79(12):1250–1259. doi:10.1001/jamaneurol.2022.3392
42. TRAILBLAZER-ALZ 4 data, presented at 15th Clinical Trials on Alzheimer’s Disease (CTAD), San Francisco November 29 – December 2, 2022..
43. Alzheon press release, Sep 20, 2022. https://alzheon.com/alzheon-reports-industry-leading-biomarker-brain-preservation-and-clinical-effects-following-12-months-of-treatment-in-phase-2-trial-of-oral-alz-801-valiltramiprosate-in-patients-with-early-alzheim/ accessed on March 7, 2023.
44. Ashton, N.J., Janelidze, S., Mattsson-Carlgren, N. et al. Differential roles of Aβ42/40, p-tau231 and p-tau217 for Alzheimer’s trial selection and disease monitoring. Nat Med 28, 2555–2562 (2022). https://doi.org/10.1038/s41591-022-02074-w
45. Portelius E, Mattsson N, Pannee J, et al. Ex vivo 18O-labeling mass spectrometry identifies a peripheral amyloid β clearance pathway. Mol Neurodegener. 2017;12(1):18. Published 2017 Feb 20. doi:10.1186/s13024-017-0152-5
46. Yanamandra K, Patel TK, Jiang H, et al. Anti-tau antibody administration increases plasma tau in transgenic mice and patients with tauopathy. Sci Transl Med. 2017;9(386):eaal2029. doi:10.1126/scitranslmed.aal2029
47. Neurofilament makes light of MS treatment monitoring. Nat Rev Neurol. 2019;15(4):188. doi:10.1038/s41582-019-0156-6
48. Lu CH, Macdonald-Wallis C, Gray E, et al. Neurofilament light chain: A prognostic biomarker in amyotrophic lateral sclerosis [published correction appears in Neurology. 2015 Sep 8;85(10):921]. Neurology. 2015;84(22):2247-2257. doi:10.1212/WNL.0000000000001642
49. https://alz-journals.onlinelibrary.wiley.com/doi/epdf/10.1002/alz.069262 accessed on March 7, 2023
50. https://precivityad.com/news/c2n-diagnostics-introduces-the-precivityad2-blood-test accessed on March 7, 2023
51. Chatterjee P, Pedrini S, Doecke JD, et al. Plasma Aβ42/40 ratio, p-tau181, GFAP, and NfL across the Alzheimer’s disease continuum: A cross-sectional and longitudinal study in the AIBL cohort [published online ahead of print, 2022 Jul 21]. Alzheimers Dement. 2022;10.1002/alz.12724. doi:10.1002/alz.12724
© Serdi 2023