
C. Fang1, P. Hernandez2, K. Liow3, E. Damiano1, H. Zetterberg4,5, K. Blennow4,5, D. Feng6, M. Chen6, M. Maccecchini1
1. Annovis Bio, Berwyn, PA, USA; 2. EZY Medical Research, Miami, FL, USA; 3. University of Hawaii, HI, USA; 4. University of Gothenburg, Sweden; 5. Clinical Neurochemistry Laboratory, Sahlgrenska University Hospital, Mölndal, Sweden; 6. TCM, NJ, USA
Corresponding Author: Cheng Fang, 1055 Westlakes Dr #300, Annovis Bio, Berwyn, PA, USA fang@annovisbio.com phone # 610-727-3987
J Prev Alz Dis 2023;1(10):25-33
Published online October 10, 2022, http://dx.doi.org/10.14283/jpad.2022.84
Abstract
Background: Previously we reported the clinical safety and pharmacological activity of buntanetap (known as Posiphen or ANVS401) in healthy volunteers and mild cognitive impaired (MCI) patients (21). The data supported continued clinical evaluation of buntanetap for treating Alzheimer’s Disease (AD). Neurodegenerative diseases such as AD and Parkinson’s disease (PD) share several pathological manifestations, including increased levels of multiple neurotoxic protein aggregates. Therefore, a treatment strategy that targets toxic species common to both disorders can potentially provide better clinical outcomes than attacking one neurotoxic protein alone. To test this hypothesis, we recently completed a clinical study in early AD and early PD participants and report the data here.
OBJECTIVES: We evaluated safety, pharmacokinetics, biomarkers, and efficacy of buntanetap in treating early AD and PD patients.
DESIGN: Double-blind, placebo-controlled, multi-center study.
SETTING: 13 sites in the US participated in this clinical trial. The registration number is NCT04524351 at ClinicalTrials.gov.
PARTICIPANTS: 14 early AD patients and 54 early PD patients.
INTERVENTION: AD patients were given either 80mg buntanetap or placebo QD. PD patients were given 5mg, 10mg, 20mg, 40mg, 80mg buntanetap or placebo QD.
MEASUREMENTS: Primary endpoint is safety and tolerability; secondary endpoint is pharmacokinetics of buntanetap in plasma; exploratory endpoints are 1) biomarkers in cerebrospinal fluid (CSF) in both AD and PD patients 2) psychometric tests specific for AD (ADAS-Cogs & WAIS coding test) or PD (MDS-UPDRS & WAIS coding test).
RESULTS: Buntanetap was safe and well tolerated. Biomarker data indicated a trend in lowering levels of neurotoxic proteins and inflammatory factors and improving axonal integrity and synaptic function in both AD and PD cohorts. Psychometric tests showed statistically significant improvements in ADAS-Cog11 and WAIS coding in AD patients and MDS-UPDRS and WAIS coding in PD patients.
CONCLUSIONS: Buntanetap is well tolerated and safe at doses up to 80mg QD in both AD and PD patients. Cmax and AUC increase with dose without evidence for a plateau up to 80mg QD. The drug shows promising evidence in exploratory biomarker and efficacy measures. Further evaluation of buntanetap in larger, longer-term clinical trials for the treatment of AD and PD are warranted.
Key words: Alzheimer’s disease (AD), Parkinson’s disease (PD), neurotoxic aggregating proteins, iron response element (IRE), iron regulatory protein (IRP).
Introduction
Alzheimer’s (AD) and Parkinson’s disease (PD) are the most common neurodegenerative diseases (1). Despite a burgeoning body of data targeting increased levels of Aβ, tau and α-synuclein (αSYN), clinical trials targeting these species have shown very limited or no efficacy. Better understanding of the pathogenesis of these diseases may enhance insights into molecular mechanisms and promote discovery of novel treatments.
Recent findings have revealed multiple neurotoxic protein aggregates in the brains of people with neurodegeneration (2). Although APP and its downstream products (Aβ oligomers, c-terminus peptide, and amyloid plaques), and tau neurofibrillary tangles have been well documented to be culprits of AD (3), other misfolded proteins are also suggested to be part of the equation. For example, in addition to Aβ and tau, increased levels of αSYN and TDP-43 have been shown to be correlated with deficits in cognitive functions in AD (2, 4). Similarly, although αSYN is the main component of Lewy bodies, TDP-43 (5), Aβ and tau (6) are also detected in PD.
While these proteins have different molecular functions, they share some common molecular pathways (7). Elevated levels of neurotoxic proteins have been shown to lead to impaired axonal transport. Axonal transport is critical for synaptic maintenance and plasticity and for the active delivery of newly synthesized compounds as well as other important materials, such as synaptic vesicle precursors, mitochondria, endosomes, autophagosomes, lysosomes etc. (8). Not surprisingly, synapse loss and dysfunction are a key feature in many neurodegenerative diseases including AD (9) and PD (10). The presence of increased levels of toxic species is also reported to impair endosomal function concurrent with failed anterograde and retrograde axonal delivery of cargoes critical for synaptic structure and function (11). Indeed, endosomal function and axonal transport are defected in both AD and PD and contribute to the disease pathology (12).
Therefore, a treatment strategy that targets toxic species common to both disorders is rational and points to the possibility that better clinical outcomes for AD and PD might be achieved by reducing the levels of toxic species found in both.
Buntanetap is an orally bioavailable small molecule derived via a biochemical synthetic pathway, which was discovered at the National Institutes of Aging (Bethesda, Maryland). Buntanetap suppresses the translation of the mRNAs of APP, tau, αSYN and other neurotoxic aggregating proteins by enhancing the binding of the atypical iron response element (IRE) in those neurotoxic proteins’ mRNAs’ 5’UTR regions to iron regulatory protein 1 (IRP1) in high iron (13-15).
By suppressing APP, tau and αSYN synthesis, we hypothesize that buntanetap normalizes the levels of these toxic proteins and re-establishes proteostasis, rescues axonal transport and endosomal function, and staves off nerve cell death and neurodegeneration. Our data in multiple animal models supports our hypothesis: we demonstrated full recovery of function in T65Dn Down Syndrome mice (16), APP/PS1 AD mice (17), PD hSNCA (either A53T or A30P) mice (18), stroke mice (19) and TBI rats (20).
We previously evaluated buntanetap’s safety in a single ascending dose (SAD), a multiple ascending dose (MAD) and in a proof of concept (POC) trial. Buntanetap was generally safe and well-tolerated. In the POC study, 5 patients with mild cognitive impairment (MCI) were given 4 x 60mg buntanetap treatment for 10 days. Buntanetap treatment effectively reduced APP and its downstream products, total tau (t-tau), phosphorylated tau (p-tau) and αSYN in patients’ CSF, supporting its inhibitory effects on these neurotoxic aggregating proteins (21).
These data warranted further study of the safety and pharmacodynamic effects of buntanetap in a larger patient population and for a longer dosing period. Further, based on the hypothesis that multiple neurotoxic protein aggregates observed in the brains of people with neurodegeneration share some common molecular pathways, we designed an exploratory study to enroll both AD and PD patients. In this study we looked at buntanetap’s safety profile (primary endpoint), pharmacokinetics (secondary endpoint), mechanism of action via biomarkers and efficacy (exploratory endpoints) in patients with early AD or early PD. Here we report our findings.
Methods
Investigational Drug
Buntanetap, (3aR)-1, 3a, 8-trimethyl-1, 2, 3, 3a, 8, 8a-hexahydropyrrolo (2, 3-b) indol-5-yl phenyl-carbamate tartrate (investigational new drug #72 654) was manufactured according to Good Manufacturing Practice (GMP) regulations (Wilmington PharmaTech, Newark, DE)
Investigational drug product and matching placebo containing a standard pharmaceutical excipient, were provided as an immediate release solid oral dosage form, prepared in hard capsule shells, manufactured in accordance with GMP regulations by Frontage Laboratories (Exton, PA).
Participants
The following inclusion criteria were applied: 1) individuals between 45 and 85 years old; 2) Female participants must be of non-childbearing potential or post-menopausal for at least 2 consecutive years or surgically sterile (bilateral tubal ligation, hysterectomy, or bilateral oophorectomy) for at least 6 months prior to screening. 3) Female participants will be given a urine pregnancy test at the screening visit for which they should test negative. 4) For AD patients, clinical dementia rating = 0.5 or 1, and MMSE score between 18 and 28. 5) For PD patients Hoehn & Yahr ≤ 3 & fulfil PD criteria by MDS-UPDRS, and MMSE score between 18 and 30. 6) General cognition and functional performance sufficiently preserved that the subject could provide written informed consent. 7) No evidence of current suicidal ideation or previous suicide attempt in the past month as evaluated in the Columbia Suicide Severity Rating Scale. 8) MRI scan within the 12 months prior to screening without evidence of infection, infarction, or other focal lesions and without clinical symptoms suggestive of intervening neurological disease. Lacunes that are not believed to contribute to the subject’s cognitive impairment are permissible. If there is no MRI available within a 12-month timeframe, then an MRI must be performed as part of the screening procedures for eligibility. 9) Are stable of permitted medications prior to screening. Exclusion criteria please see supplementary methods. Written informed consent was obtained from all participants and the study protocol was approved by Institutional Review Boards (IRBs). Details of exclusion criteria are provided in Supplementary Method 1.
Randomization
Subjects who signed an informed consent and met screening eligibility requirements were randomly assigned to the active and placebo treatment groups (10 randomized to buntanetap and 5 to Placebo for AD and PD, respectively) in Part 1. In Part 2, 40 Early PD patients were randomized to 10 patients each to one of the 4 different dose levels of buntanetap (5, 10, 20 or 40mg). This is a quadruple (participants, care providers, investigators, and outcomes assessors) blinded study.
Trial Design
A total of 68 AD and PD patients were treated for 25±2 days. All patients consented to voluntarily participate in the clinical trial. These sample sizes were determined to provide enough data to characterize the safety, tolerability, and PK of buntanetap as well as adequately supporting potential dose proportionality analyses. This was an exploratory study, beyond safety and pharmacokinetics, biomarkers focusing on target engagement, pathway engagement and functional/cognitive measures were also investigated.
Part 1 of the study included 14 early AD and 14 early PD patients treated with either 80mg buntanetap QD or placebo for 25±2 days. Part 2 of the study involved 40 early PD patients treated with 5mg, 10mg, 20mg or 40mg buntanetap QD for 25±2 days. In both parts of the study, patients’ blood and CSF samples were collected at baseline before treatment and for 6 hours (hrs) after 25±2 days treatment. MDS-UPDRS and WAIS coding test were performed at baseline before treatment and after 25±2 days treatment in PD patients and ADAS-Cog11 and WAIS coding test were performed in AD patients.
Standards Deuterated buntanetap, N8- and N1- norbuntanetap metabolites were synthesized by Chemtos (Round Rock, TX) to >99% purity.
Biomarker Assays
Biomarkers were analyzed by Clinical Neurochemistry Laboratory of Sahlgrenska University Hospital (Mölndal, Sweden).
CSF sAPPα and sAPPβ, YKL-40 concentrations were measured using enzyme-linked immunosorbent assays (ELISAs), as described by the kit manufacturers (IBL International GmbH, Hamburg, Germany) (R&D Systems, Minneapolis, MN). CSF Aβ40, Aβ42, t-tau, and p-tau concentrations were measured using Lumipulse (Fujirebio, Ghent, Belgium) (22). CSF αSYN concentrations were determined using an immunoassay with electrochemiluminescence detection (Meso Scale Discovery, Rockville, MD). CSF sTREM2 concentrations were assessed using an in-house Meso Scale Discovery assay (23). CSF GFAP and NFL concentrations were measured using an in-house ELISA (24).
Buntanetap Pharmacokinetics
To determine the concentration of buntanetap in human plasma, a high-performance liquid chromatographic mass spectrometric detection method was validated at Charles River Laboratories (Montreal ULC) using deuterated buntanetap as standard for the concentration assessments. The bioanalytical method was performed in accordance with current FDA, Industry Guidelines and as well as OECD Principles. The determination of buntanetap in human plasma was performed using an assay range of 0.100 to 150 ng/mL. The sample analysis was conducted in accordance with current GCP and GLP principles, and the results were presented for pharmacokinetic profiling. Details see Supplementary Method 2.
Statistical Analysis
Pharmacokinetics (PK) endpoints were based on the assessments of buntanetap levels in plasma. Samples were collected at pre-dosing (0hr) and at 1, 2, 3, 4, 5, and 6 hrs for baseline and Day 25±2 (Confinement) visit. Change from baseline in the PD endpoints were analyzed via MMRM (Mixed Model for Repeat Measures) methods. The analysis model included treatment, time, and treatment-by-time interaction as the fixed effects. The compound symmetry covariance structure was assumed in the analyses. The estimated overall treatment effects, the treatment effects at all post treatment timepoints, the treatment differences, and the two-sided 95% confidence intervals for the estimated effects were reported.
Statistical analysis for treatment changes (the change from baseline (Day 25 – Day 0)) and for between treatment changes (differences between treatment and placebo) in efficacy endpoints, including ADAS-Cog11, WAIS coding test and MDS-UPDRS, were analyzed using an analysis of variance (ANOVA) model. The model included treatment as fixed effects. Data were presented as Mean ± Standard Error.
Results
Participants’ flow, adherence to interventions, and background characteristics
This trial was exploratory in nature. In Part 1 of the trial, we recruited both early AD and early PD patients to be treated with either 80mg QD buntanetap (the highest safe dose based on our SAD study) or placebo. Our data in animal studies suggest that buntanetap is efficacious at lower doses and we tested that with a dose-finding Part 2 study in PD patients. Although Part 1 and 2 are conceptually independent, they were done sequentially with overlap in the recruitment window. We had a faster recruiting rate in PD patients than AD patients. So as soon as the 15 PD patients for Part 1 were recruited, we started recruiting PD patients for Part 2, during which the recruitment for Part 1 AD was still ongoing and all data were blinded. Therefore, Part 1 PD data can be combined with Part 2 PD data as one continuous study for dose response.
A total of 101 AD and PD patients were screened. 17 AD patients and 58 PD patients were enrolled and randomized. Figure 1 shows the flow from screening to end of study participants. AD patients were randomized into 80mg QD buntanetap and placebo and PD patients were randomized into 5mg, 10mg, 20mg, 40mg, 80mg QD buntanetap and placebo. A total of 64 were assigned to the intervention groups (11 AD patients and 53 PD patients) and 11 were assigned to the control group (6 AD patients and 5 PD patients). After withdrawal from consent, discontinuation due to protocol violation (non-adherence to study drug) and Covid infection, 69 patients (92%) finished the study (Figure 1). Baseline characteristics were balanced between the intervention and control groups, as there were no significant differences in age, sex, comorbidities, cognitive scores, and H&Y scores in PD population. We recruited at total of 36% of patients from ethnic and racial minority groups (Demographic data see Supplementary Table 1).
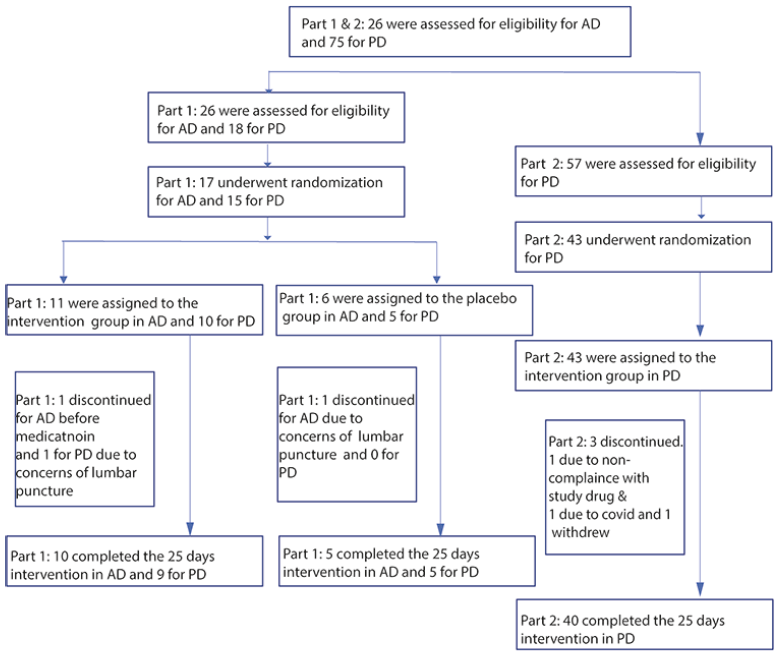
Figure 1. Enrolment, randomization, and completion of AD and PD trials
Buntanetap was safe and well-tolerated in both AD and PD patients
Safety was the primary endpoint of the study, and it was measured by adverse events (AEs), concomitant medication monitoring, 12-lead ECGs, clinical laboratory testing, vital signs assessments, and physical examinations.
Buntanetap was safe and well-tolerated in both patient populations. The majority of AEs were attributed to the study procedures (lumbar puncture). There were no clinically significant findings identified in the vital sign measurements or physical examinations. A single AE was noted for a Grade 1 QT prolongation in a patient receiving buntanetap at the confinement visit admission, which resolved and was considered non-clinically significant based on medical history.
There was no evidence of treatment-related clinical laboratory testing abnormalities which were considered clinically significant by the investigators. In AD patients, a single report of elevated liver function tests (AST, Alk Phos, Bili) was noted in a patient receiving buntanetap at the study confinement admission visit, all of which were resolved and were not considered related to study drug. There were no SAEs, or AEs resulting in drug withdrawal or study discontinuation. In PD patients, there were no trends relative to buntanetap dose increase. Treatment-emergent adverse events (TEAEs) noted as related (or possibly related) to study drug were headache, erythema, movement disorder, and muscle spasms. All were mild (Grade 1). (Safety table see Supplementary Table 2).
Pharmacokinetics of buntanetap in AD and PD patients
In this study, a total of 50 PD patients were given either 5, 10, 20, 40 or 80mg buntanetap QD and 10 AD patients were given 80mg buntanetap QD for 25±2 days. Calculated PK parameters for buntanetap in plasma of these AD and PD patients were similar to each other. They were also similar to the PK parameters we have from prior studies in healthy volunteers (SAD and MAD), and in MCI patients in POC study. In this study, overall, mean Cmax and AUC values for plasma concentration data increased in an approximately dose-proportional manner as the dose increased from 5 mg to 80 mg. In plasma, buntanetap mean Tmax was 1.22-1.89 hrs (Table 1).
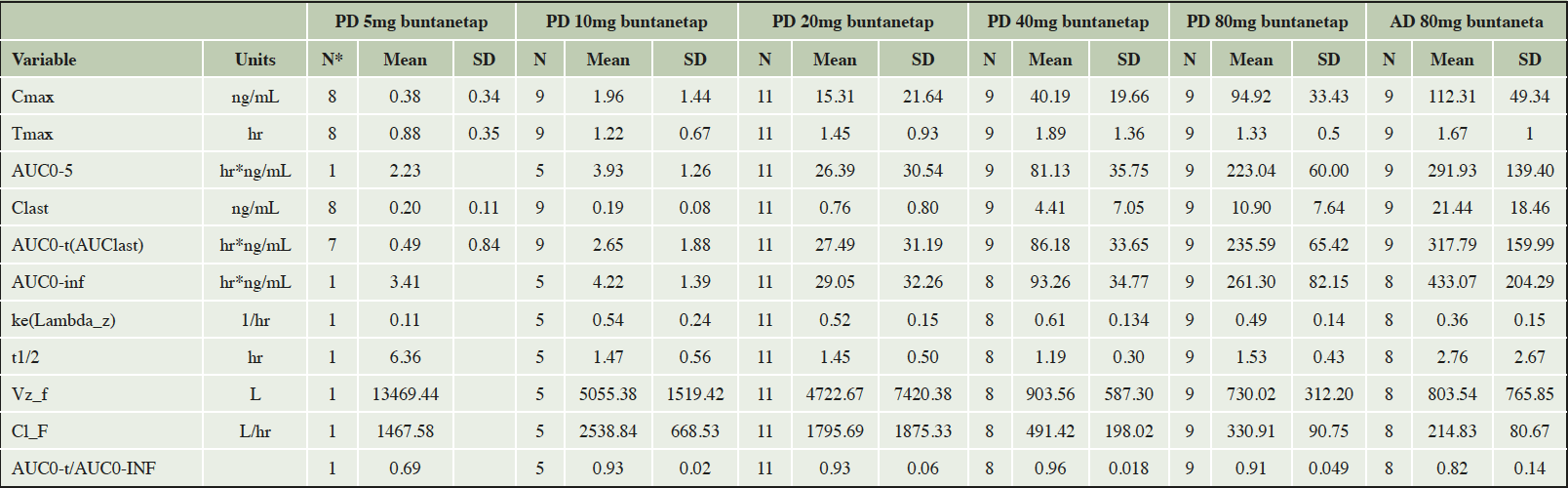
Table 1. Pharmacokinetics of buntanetap in AD and PD patients
*Due to the low plasma concentration, PK from 5mg buntanetap QD group cannot be reliably detected.
The T1/2 from our SAD, MAD, and MCI patients are all very consistent and are about 4-5hrs in plasma[21]. In this double study, due to short PK sample collection time (0-6 hrs instead of 0-12 hrs), the T1/2 of both AD and PD patients treated with buntanetap are shorter. When calculating 60 mg QD PK data (0-12hrs) in mild AD patients from our ADCS study (unpublished data), the predicted T1/2 is 3.84hr, consistent with what we observed before and further supporting that the shorter T1/2 we observed here is simply due to sample collection over 6 hrs only. It is a well-known issue that if the span of time over which a half-life is estimated is shorter than the half-life, the results of that estimation are not robust (25). This is because the slope of the linear decay (from which the elimination rate constant is determined) is defined by a limited duration and a very limited number of samples. We will validate the T1/2 in AD and PD patients in future clinical trials with longer PK sample collection time.
Importantly, although we didn’t measure buntanetap level in CSF in this study, our previous study shows that buntanetap’s T1/2 in CSF of MCI patients was much longer than its T1/2 in plasma, >12 h versus approximately 4-5 h, respectively (21). That’s why we choose to dose patients once per day in this study.
Buntanetap statistically improved AD patients’ cognition
We evaluated buntanetap’s effect in AD patients’ cognition using ADAS-Cog11 and WAIS coding test. The ADAS-Cog11 is one of the most frequently used tests to measure cognition in research studies and clinical trials. The placebo group improved ADAS-Cog 11 slightly, 1.1±2.63 points. However, buntanetap treatment improved patients’ ADAS-Cog11 score by 4.40±2.04 points, a statistically significant improvement compared to their baseline and 3.3±3.32 points better compared to placebo (Figure 2A).
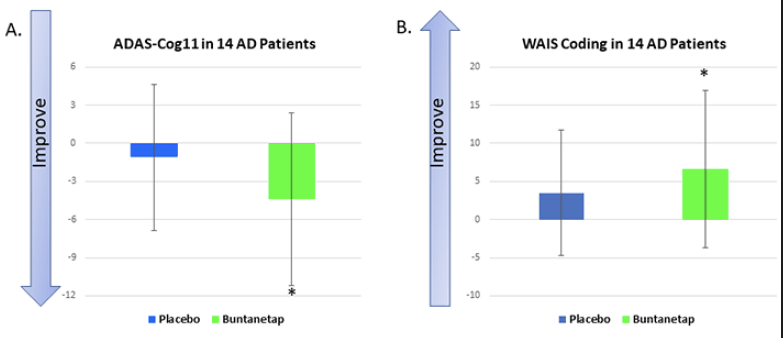
Figure 2. Cognitive functions were measured in 14 AD patients
In each test, statistical analysis was done to compare placebo post-treatment to its own baseline, buntanetap post-treatment to its own baseline, and buntanetap post-treatment to placebo post-treatment. A. From baseline to 25 days in the buntanetap-treated group, ADAS-Cog11 improved by 4.4 points, a statistically significant improvement over its own baseline. B. The WAIS coding test measures speed in movement and thinking. Buntanetap-treated AD patients showed a 6.6-point improvement in coding after 25±2 days, a significant improvement over its own baseline. *P<0.05
Within ADAS-Cog11, we also examined two different subsets that are potentially more responsive than ADAS-Cog11 to treatment effects, ADAS-Cog3 and ADAS-Cog6 (26). Buntanetap-treated group showed a trend of improvement in both subsets. We also measured ADAS-Cog14, an expanded version of the ADAS-Cog11 that includes all ADAS-Cog11 items as well as three additional items to assess delayed word recall, executive function, and number cancellation. Similarly, buntanetap-treated group showed a trend of improved test performance (Supplementary Figure 1).
Besides ADAS-Cogs, we also measured WAIS coding test. WAIS coding measures visual-motor dexterity, associative nonverbal learning, and nonverbal short-term memory. Speed has been shown to play a primary role in WAIS coding scores and memory plays a moderate but genuine secondary role as well (27). Fine-motor dexterity, speed, accuracy, and ability to manipulate a pencil contribute to task success; perceptual organization is also important. A similar coding test has been reported to provide particularly good discrimination (AUC: .785; 95% CI: .72-.85) to controls (CDR 0) from those with CDR 0.5 (which includes MCI and very mild dementia) (28). Buntanetap-treated AD patients showed a 6.6±3.04 points improvement after 25±2 days, a statistically significant improvement from baseline (Figure 2B). We also tested MMSE and CDR sum of boxes in AD patients and, while we saw a trend, we didn’t see any statistical differences (Supplementary Figure 2).
Buntanetap statistically improved PD patients’ mobility shown by MDS-UPDRS (both Part III and total score) and WAIS coding score
Part III of the MDS-UPDRS measures the motor functions and has long been used as a standard to assess drugs’ effects on patients’ mobility. Buntanetap at all doses improved patients’ Part III scores. We observed the best improvement after buntanetap treatment with 10mg and 20 mg QD, statistically significant improvement compared to baseline (Figure 3A).
When comparing the total scores of MDS-UPDRS, a gold-standard for assessing a drug’s effects on PD patients’ disease progress, we again observed the best improvement with 10mg and 20mg buntanetap treatment. Both 10 and 20mg buntanetap treated patients score higher than placebo and show significant improvement when compared with their own baseline (Figure 3B).
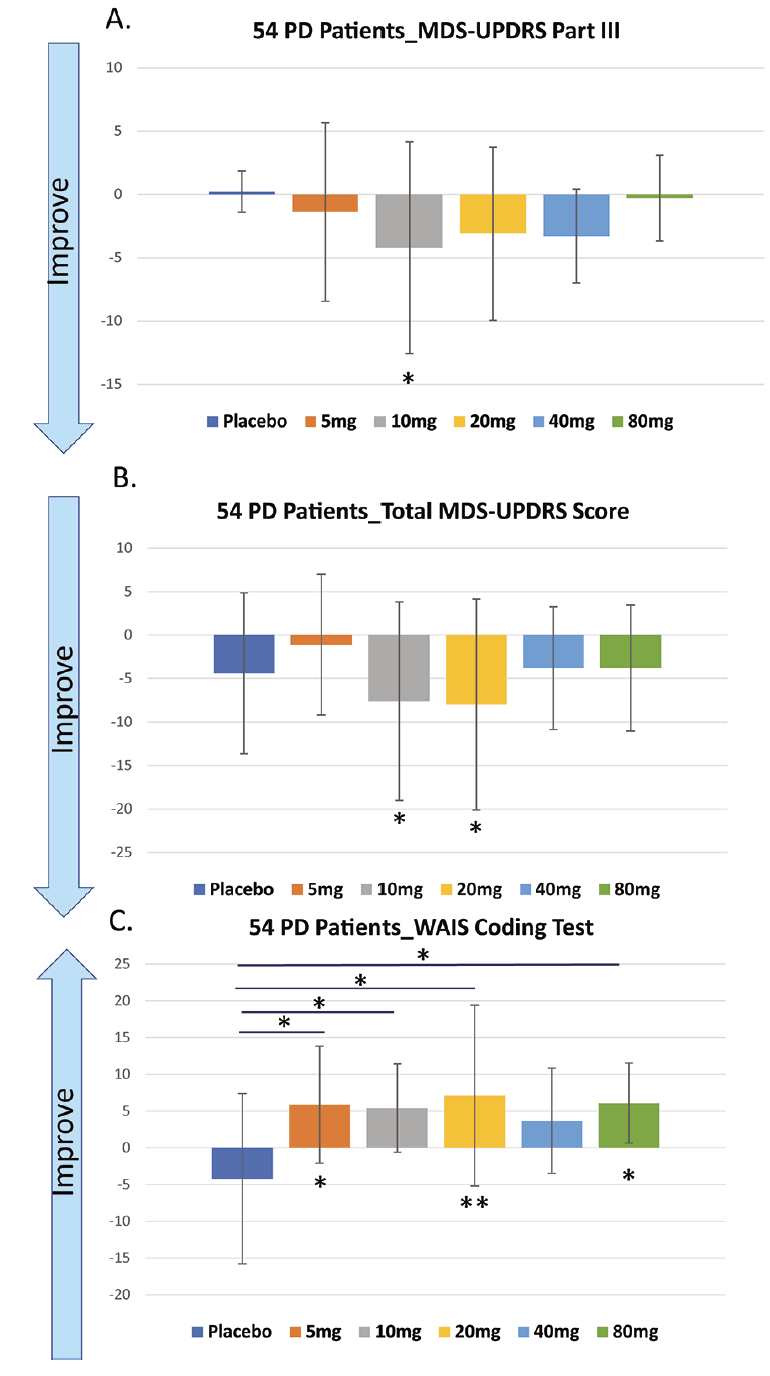
Figure 3. Mobility was measured in 54 PD patients
In each test, statistical analysis was done to compare placebo post-treatment to its own baseline, buntanetap post-treatment to its own baseline, and buntanetap post- treatment to placebo post-treatment A. From baseline to 25±2 days, buntanetap improved patients MDS-UPDRS Part III score, especially at 10mg QD. B.. Buntanetap improved patients total MDS-UPDRS score. Both 10mg and 20mg QD group showed significant improvement compared to baseline. C. From baseline to 25±2 days, even 5mg QD buntanetap significantly improved patients WAIS coding score, both compared to baseline and to placebo Statistical analysis was also done comparing placebo after treatment vs placebo baseline. * P<0.05; ** P<0.01
Buntanetap, even at 5mg QD statistically significantly improved patients’ WAIS coding score both compared to baseline and to placebo. This effect has been observed with almost all dosages with 20mg showing the best statistically significant result both compared to its own baseline and to placebo (Figure 3C).
Buntanetap showed a trend of reducing neurotoxic proteins and inflammation and increasing axonal integrity and synaptic function in both AD and PD patient.
Our analyses of biomarkers assume that each patient’s biomarker level is 100% at baseline; we then measured the levels of the same biomarkers at 25±2 days to calculate the percentage changes comparing to baseline. Although the sample size was not powered to see any statistical significance in biomarkers, we were encouraged to see a trend in the right direction in all biomarkers measured. 80mg QD buntanetap treatment reduced Aβ40, sAPPα, sAPPβ, t-tau and p-tau, common neurotoxic proteins involved in AD pathology while Aβ42 levels in buntanetap-treated group slightly increased (Figure 4A). These data agree with our outcomes from in vitro studies, different animal models and MCI patients and support buntanetap’s MOA as inhibiting neurotoxic aggregating proteins.
Inflammation is a central mechanism of both AD and PD disease. We analyzed four inflammatory markers. GFAP is a known marker for astroglia injury[29]. Triggering receptor expressed on myeloid cells 2 (TREM2) is an innate immune receptor expressed by microglia. Its cleaved fragments, soluble TREM2 (sTREM2), can be measured in the CSF as a biomarker for inflammation[30]. YKL-40 is a glycoprotein, and its level is elevated in the brain and CSF in several neurological and neurodegenerative diseases associated with inflammatory processes (31). Complement C3 is part of the innate immune system involved in clearance of pathogens and damaged cells, is expressed, and secreted by microglia and astrocytes and can participate in synapse removal. (32). Here we showed that compared with placebo-treated group, buntanetap-treated group saw a reduction of sTREM2, GFAP and Complement C3 levels after 25±2 days (Figure 4B). These data suggest a potential reduction of inflammation in buntanetap treated AD patients.
Then we set out to examine buntanetap’s effects on neuronal functions. Neurofilament light (NFL) is a sensitive biomarker of neuroaxonal damage. NFL has been widely adopted to reflect disease severity in AD (33). In buntanetap-treated group, there was less NFL increase comparing to placebo group, suggesting a potentially improved axonal integrity. We also measured neurogranin (NG). NG is a calmodulin-binding protein expressed primarily in the brain, particularly in dendritic spines. Recent studies have shown that NG is involved in the plasticity and regeneration of synapse mediated by the calcium- and calmodulin-signaling pathways. It therefore is a biomarker for synaptic integrity and functions (34). 25±2 days later CSF level of NG in placebo group increased 16.56±4.90% while in buntanetap treatment group, NG CSF only increased 3.81±3.65%, suggesting buntanetap potentially reduced the damage to synaptic integrity/ functions (Figure 4C).
Similarly, the 14 PD patients given 80 mg QD buntanetap had reduced αSYN CSF levels compared to placebo (Figure 4D). We also analyzed the same inflammatory markers in CSF and buntanetap treatment reduced all four inflammatory markers (sTREM, GFAP, Complement C3 and YKL-40) after 25±2 days. These data suggest a reduction of inflammation in buntanetap treated PD patients (Figure 4E). NFL has also been widely adopted to reflect disease severity in PD (35). CSF NFL level in in placebo group increased 8.97±5.42% while it decreased 0.61±3.61% in buntanetap treatment group, suggesting potentially improved axonal integrity (Figure 4F). NG level also decreased in buntanetap treatment group, suggesting potentially improved synaptic functions comparing to placebo group (Figure 4F).
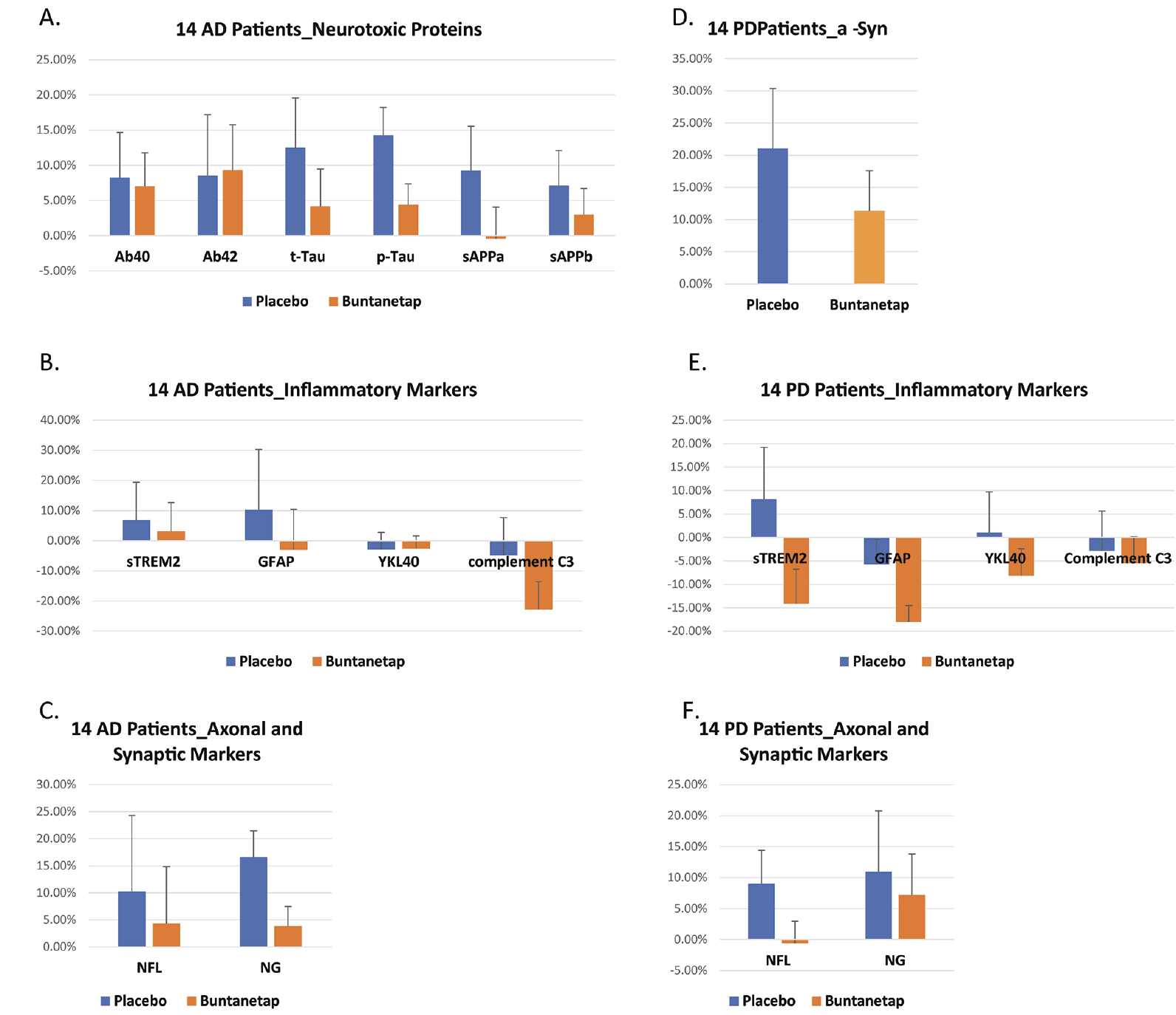
Figure 4. CSF biomarkers were measured in 14 AD patients and 14 PD patients (80mg vs Placebo)
In each test, statistical analysis was done to compare placebo post-treatment to its own baseline, buntanetap post-treatment to its own baseline, and buntanetap posttreatment to placebo post-treatment. Although none of the comparison reached statistical significance, compared to placebo, buntanetap treatment showed a trend to A. reduce AD patients’ sAPPα, sAPPb, Ab40, t-Tau and p-Tau. B. reduce AD patients’ inflammatory markers sTREM2, GFAP and Complement C3 levels with YKL-40 levels unchanged. C. reduce AD patients’ CSF NFL and neurogranin (NG) levels. D. reduce a-Syn in PD patients. E. reduce PD patients’ inflammatory markers sTREM2, GFAP, YKL-40 and Complement C3. F. reduce PD patients’ NFL levels while neurogranin (NG) levels remained the same. Y axis is the percentage difference (%) in CSF at 25±2 days comparing to baseline.
Discussion
Our data suggest that buntanetap was safe and well tolerated in AD and PD patients. With the caveat that this study was not powered to have statistically significant results in biomarkers and didn’t produce statistically significant results in biomarkers, we were encouraged to see that biomarker data in both AD and PD patients show the right trend. This suggests that buntanetap potentially lowered the levels of neurotoxic proteins, improved neuronal function and decreased inflammation, consistent with pre-clinical data in animal models. Despite the small sample size, to our surprise buntanetap improved cognition in AD patients and motor function in PD patients, both statistically significantly.
Although we still do not fully understand the disease mechanisms of AD and PD, recent findings have pointed to an unarguable involvement of multiple neurotoxic proteins (2, 4). This can help explain why the drugs focusing on removing single toxic protein have so far shown no or minimal effects.
Buntanetap lowers the levels of more than one neurotoxic protein (APP, αSYN, tau, TDP43, Huntingtin protein etc.). These mRNAs contain the same conserved IRE in their 5’ UTR regions and their mRNA translation is regulated by binding to the same IRP1 (36). In low iron, IRP1 binds to these IREs and thus prevents the mRNAs from going to the ribosome and being translated. In high iron, IRP1 releases the IREs and these mRNAs will then bind to the ribosome and start translation (37). In the same high iron, buntanetap keeps the IREs bound to IRP1, thus the mRNAs are not translated and proteins not overexpressed. Cellular iron levels are known to be elevated in multiple neurodegenerative diseases (38) and cause the over-expression of these neurotoxic proteins (39). Therefore, regulating IRE-IRP1 binding provides a novel mechanism of action to regulate the expression of these neurotoxic proteins.
Importantly, buntanetap specifically binds to the atypical IRE loop/ IRP1 complex with very high affinity – IC50 3.2 nM. It does not affect the typical, canonical IRE loops present in the mRNAs of iron metabolism proteins. The specific interaction lowers the translation level of neurotoxic proteins without affecting the iron regulatory mechanisms of the cell (13, 39).
Our PK study shows that Cmax and AUC both increase without evidence of a plateau up to 80mg QD. However, in PD patients, 10 and 20mg QD groups showed best efficacy results in both MDS-UPDRS and WAIS coding test. Our animal studies suggest the efficacious dose of buntanetap is around 150ng/gram in the brain, similar to the extrapolated concentration from 10 and 20mg QD in humans. We will further test this hypothesis with a dose finding study in AD patients.
We hypothesized that by inhibiting multiple neurotoxic proteins, we would rescue neuronal function. This hypothesis has been supported by our data in animal models (16, 19) and presently supported by our current clinical research data in both AD and PD patients.
Given the short duration of this clinical trial, it was quite unexpected to see buntanetap improving patient’s cognition and function. In line with recent literature (40), we hypothesize that the effects on cognition and motor function after such a short treatment are due to buntanetap’s ability to restore neuronal function by improving axonal integrity, improving axonal transport, improving synaptic transmission, and reducing inflammation. These fast-acting effects suggest that buntanetap can function as a symptomatic drug to give patients quick relief of their neurological symptoms. However, based on our animal studies, we believe that buntanetap can also function as a disease-modifying drug in both AD and PD patients as it will continue to reduce the levels of neurotoxic aggregating proteins, preserve neuronal integrity and prevent neuronal loss in the long run and thus prevent patients from losing their cognition and mobility. We plan to test this hypothesis further in our future clinical trials (phase 3 in PD and phase 2/3 in AD) with larger patient populations and longer treatment times.
Conflict of interest: Cheng Fang, Eve Damiano & Maria Maccecchini are employed by Annovis Bio. Priscilla Hernandez & Kore Liow received funding from Annovis Bio for the conduct of the clinical trial, Henrik Zetterberg and Kaj Blennow received funding from Annovis Bio for biomarker analysis and Michael Chen and David Feng received funding from Annovis Bio for statistical analysis.
Ethics approval: The human studies were approved by Advarra Institutional Review Board (IRB) (Columbia, MD, USA) and IRB boards of University of Pennsylvania and University of Texas. Participants provided written consent to participate with IRB approved informed consent forms.
Acknowledgements: The authors would like to thank the participants and their study partners, without whom this study would not have been possible.
Funding: This study was supported by Annovis Bio.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. The Challenge of Neurodegenerative Diseases, in Harvard NeuroDiscovery Center.
2. Peterson, R., How early can we diagnose Alzheimer disease. Neurology, 2017.
3. Breijyeh Z, K.R., Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules, 2020.
4. Schneider J, A.Z., Leurgans S, Bennett D, The Neuropathology of Probable Alzheimer. Ann Neurol, 2009.
5. Nakashima-Yasuda H, U.K., Robinson J, Xie SX, Hurtig H, Duda JE, Arnold SE, Siderowf A, Grossman M, Leverenz JB, Woltjer R, Lopez OL, Hamilton R, Tsuang DW, Galasko D, Masliah E, Kaye J, Clark CM, Montine TJ, Lee VM, Trojanowski JQ, Comorbidit of TDP-43 proteinopathy in Lewy boday related diseases. Acta Neuropathol, 2007.
6. Lim EW, A.D., Ffytche D, Chaudhuri KR, Kings Parcog group & MDS Nonmotor study group, Amyloid-β and Parkinson’s disease. J Neurol, 2019.
7. Alami N, S.R., Carrasco M, Shaw C, Taylor P, Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron, 2014.
8. Guedes-Dias P, H.E., axonal transport: driving synaptic function. science, 2019.
9. Selkoe, D., Alzheimer’s disease is a synaptic failure. Science, 2002: p. 789-791.
10. Picconi B, P.G., Calabresi P, Synaptic dysfunction in Parkinson’s disease. Adv Exp Med Biol., 2012.
11. Stamer K, V.R., Thies E, Mandelkow E, Mandelkow EM, Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol, 2002: p. 1051-63.
12. Chung CY, K.J., Siddiqi H, Isacson O, Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J Neurosci, 2009.
13. Chen XQ, B.C., Carpio RV, Rogers J and Maccecchini M, Posiphen Reduces the Levels of Huntingtin Protein through translation suppression. Pharmaceutics, 2021.
14. Rogers JT, M.S., Cantuti-Castelvetri I, Smith DH, Huang X, Bandyopadhyay S, Cahill CM, Maccecchini ML, Lahiri DK, Greig NH., The alpha-synuclein 5’ untranslated region targeted translation blockers: anti-alpha synuclein efficacy of cardiac glycosides and posiphen. 2011.
15. Cho HH, C.C., Vanderburg CR, Scherzer CR, Wang B, Huang X, Rogers JT., Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein-1. 2010.
16. Chen XQ, S.A., Pearn M, Maccecchini M, Mobley W, Targeting increased levels of APP in Down syndrome: Posiphen-mediated reductions in APP and its products reverse endosomal phenotypes in the Ts65Dn mouse model. Alzheimer’s & dementia, 2020.
17. Teich AF, S.E., Barnwell E, Arancio O, Maccecchini M, Translational inhibition of APP by Posiphen: Efficacy, pharmacodynamics, and pharmacokinetics in the APP/PS1 mouse. Alzheimer’s & dementia 2018.
18. Kuo YM, N.E., Nussbaum R, Rogers J and Maccecchini M, Translational inhibition of α-synuclein by Posiphen normalizes distal colon motility in transgenic Parkinson mice. Am J Neurodegener Dis, 2019: p. 1-15.
19. Turcato F, K.P., Barnett A, Jin YM, Greig N and Luo Y, Sequential combined Treatment of Pifithrin-a and posiphen enhances neurogenesis and functional recovery after stroke. Cell Transplantation 2018: p. 607-621.
20. Hatami A, M.K., Sutton R, Ghavim S, Dutta G, Maccecchini M, Chesselet MF, Posiphen reverses behavioral deficits and neuropathology in a rat model of mild traumatic brain injury. 2016, Military Health System Research Symposium.
21. Maccecchini M, C.M., Pan C, Zetterberg H, Greig N, Posiphen as a candidate drug to lower CSF amyloid precursor protein, amyloid-b peptide and tau levels: target engagement, tolerability and pharmacokinetics in humans. JNNP, 2012.
22. Goborm J, P.L., Rosa-Neto P, Vyhnalek M, Gauthier S, Zetterberg H, Sheardova K, Hort J, Blennow K, Validation of the LUMIPULSE automated immunoassay for the measurement of core AD biomarkers in cerebrospinal fluid. 2021.
23. Ashton N, S.-C.M., Heslegrave A, Blennow K, Hodges A, Zetterberg H, Plasma levels of soluble TREM2 and neurofilament light chain in TREM2 rare variant carriers. 2019.
24. Rosengren L, W.C., Hagberg L, A sensitive ELISA for glial fibrillary acidic protein: application in CSF of adults. 1994.
25. Rosenbaum, S., in Basid Pharmacokinetics and Pharmacodynamics. 2016, John Wiley & Sons: Hoboken, NJ. p. 102.
26. Kueper J, S.M., Montero-OOdasso M, The Alzheimer’s disease assessment scale-cognitive subscale (ADAS-Cog): modifications and responsiveness in pre-dementia populations. A narrative review. J Alzheimers Dis., 2018.
27. Joy S, K.E., Fein D, Speed and memory in the WAIS-III digital symbol-coding subtest across the adult lifespan. neuropsychology, 2004: p. 759-767.
28. Galvin JE., T.M., Moore C., Chrisphonte S., The number symbol coding task: a brief measure of executive function to detect dementia and cognitive impairment. Plos One, 2020.
29. Teitsdottir U, J.M., Lund S, Darreh-Shori T, Snaedal J, Petersen P, Association of glial and neuronal degeneration markers with Alzheimer’s disease cerebrospinal fluid profile and cognitive functions. Alzheimer’s research & therapy, 2020.
30. Ulland T, C.M., TREM2-a key player in microglial biology and Alzheimer disease. Nature reviews neurology, 2018: p. 667-675.
31. Muszynski P, G.M., Kulakowska A, Mroczko B, YKL-40 as a potential biomarker and a possible target in therapeutic strategies of Alzheimer’s disease. Curr. Neuropharmacol., 2017: p. 906-917.
32. Wu T, D.B., Gandham V, Carano R, Sheng M, Hanson J, Complement C3 is activated in human AD brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Reports, 2018.
33. Lewczuk P, E.N., Andreasson U, Blennow K, Zetterberg H, Plasma neurofilament light as a potential biomarker of neurodegeneration in Alzheimer’s disease. Alzheimer’s research & therapy, 2018.
34. Liu W, L.H., He X, Chen L, Dai Y, Jia W, Xue X, Tao J, Chen L, Neurogranin as a cognitive biomarker in cerebrospinal fluid and blood exosomes for Alzheimer’s disease and mild cognitive impairment. Nature, 2020.
35. Backstrom D, L.J., Mo S, Riklund K, Zetterberg H, Blennow K, Forsgren L, Lenfeldt N, NFL as a biomarker for neurodegeneration and survival in Parkinson disease. Neurology, 2020.
36. Anderson C, S.M., Eisenstein R, Leibold E, Mammalian iron metabolism and its control by iron regulatory proteins. Biochem Biophys Acta, 2012.
37. Cho HH, C.C., Vanderburg C, Rogers J, Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein 1. J Biol Chem, 2010.
38. Lane D, A.S., Bush A, Iron and Alzheimer’s disease: an update on emerging mechanisms. Journal of Alzheimer’s Disease, 2018.
39. Bandyopadhay S, C.C., Balleidier A, Huang C, Rogers J, Novel 5’ Untranslatied region directed blockers of iron-regulatory protein-1 dependent amyloid precursor protein translation: implications for down syndrom and alzheimer’s disease. Plos One, 2013.
40. Chen XQ, M.W., Exploring the Pathogenesis of Alzheimer Disease in Basal Forebrain Cholinergic Neurons: Converging Insights from Althernative Hypotheses. Frontier in Neuroscience, 2019.