
E. Siemers1, J. Hitchcock1, K. Sundell1, R. Dean1, J. Jerecic2, E. Cline2, K. Iverson2, J. Moore2, C. Edgar3, R. Manber4, N. Fuin4, T. Poppe4, R. Barton1
1. Acumen Pharmaceuticals, Inc. – Carmel, IN, USA; 2. Acumen Pharmaceuticals, Inc. – Charlottesville, VA, USA; 3. Cogstate Ltd. – Melbourne, Australia; 4. Ixico Plc – London, UK.
Corresponding Author: Eric Siemers, Acumen Pharmaceuticals, Inc., 11711 N. Meridian St., Ste. 310, Carmel, IN 46032, USA, esiemers@acumenpharm.com
J Prev Alz Dis 2023;1(10):19-24
Published online October 26, 2022, http://dx.doi.org/10.14283/jpad.2022.93
Abstract
Background: Alzheimer’s disease is a large and growing unmet medical need. Clinical trial designs need to assess disease-related outcomes earlier to accelerate the development of better treatments for Alzheimer’s disease. ACU193 is a monoclonal antibody that selectively targets amyloid β oligomers, thought to be the most toxic species of Aβ that accumulates early in AD and contributes to downstream pathological effects. Nonclinical data indicate that ACU193 can reduce the toxic effects of amyloid β oligomers. ACU193 is currently being investigated in a phase 1 clinical trial designed with the properties described in this report. This phase 1 trial is designed to provide data to enable a go/no-go decision regarding the initiation of a subsequent phase 2/3 study.
Objectives: To design a phase 1 study that assesses target engagement and incorporates novel measures to support more rapid development of a potential disease-modifying treatment for Alzheimer’s disease.
Design: The INTERCEPT-AD trial for ACU193 is an ongoing randomized, placebo-controlled phase 1a/b study that assesses safety, tolerability, pharmacokinetics, target engagement, clinical measures, and several Alzheimer’s disease biomarkers, including novel digital and imaging biomarkers.
Setting: For INTERCEPT-AD, brief inpatient stays for patients in the single ascending dose portion of the study, with the remainder of the evaluations being performed as outpatients at multiple clinical trial sites in the U.S.
Participants: Patients with early Alzheimer’s disease (mild cognitive impairment or mild dementia with a positive florbetapir positron emission tomography scan).
Intervention: ACU193 administered intravenously at doses of 2– 60 mg/kg.
Measurements: Safety assessments including magnetic resonance imaging for the presence of amyloid-related imaging abnormalities, clinical assessments for Alzheimer’s disease including the Alzheimer’s Disease Rating Scale-cognition and Clinical Dementia Rating scale, pharmacokinetics, a measure of target engagement, and digital and imaging biomarkers, including a computerized cognitive test battery and a measure of cerebral blood flow using arterial spin labelling magnetic resonance imaging.
Results: A phase 1 study design was developed for ACU193 that allows collection of data that will enable a go/no-go decision for initiation of a subsequent adaptive phase 2/3 study.
Conclusions: A phase 1a/b trial and an overall clinical development plan for an Alzheimer’s disease treatment can be designed that maintains patient safety, allows informed decision-making, and achieves an accelerated timeline by using novel biomarkers and adaptive study designs.
Key words: Alzheimer’s disease therapeutics, monoclonal antibody, Aβ oligomers, phase 1 trial design, computerized cognitive battery, arterial spin labeling.
Introduction
The search for disease-modifying therapies for Alzheimer’s disease (AD) has extended over at least two decades, and has been challenging, but recent results suggest that treatments with a clinically meaningful slowing of disease progression are on the horizon (1). While drug mechanisms that are not directly related to amyloid beta (Aβ) are of great interest, a large amount of genetic and biomarker data suggest that Aβ is important for AD pathology. The relationship between Aβ and AD is complex in that Aβ may exist as soluble monomers, soluble oligomers, soluble or insoluble protofibrils, insoluble fibrils, or insoluble amyloid plaques.
Recent publications have suggested that monoclonal antibodies specifically targeting amyloid plaque, e.g., aducanumab and donanemab, may slow cognitive decline of AD (2, 3). The trials for both aducanumab and donanemab showed a substantial incidence of a treatment-emergent adverse event known as amyloid-related imaging abnormalities-edema (ARIA-E), which will need to be weighed against any clinical efficacy shown in subsequent trials. Lecanemab, a monoclonal antibody designed to target Aβ soluble protofibrils (4), has been shown in a phase 2 clinical trial also to lower plaque load and to have a risk of ARIA (5), and thus likely has some binding to amyloid plaque. Phase 2 data also suggests that lecanemab slows cognitive decline in patients with early AD.
A substantial body of evidence has also accrued over several years suggesting that soluble Aβ oligomers (sAβOs) represent the most toxic species of Aβ, accumulating early in AD and driving downstream pathology of AD, including the aberrant phosphorylation of tau (p-tau) and neurodegeneration (6), as well as cognitive decline.
Background and rationale for ACU193 development
ACU193 is the first monoclonal antibody to enter clinical trials that was designed to bind to sAβOs with high selectivity. As reported elsewhere (7, 8), ACU193 binds with over 600-fold selectivity for sAβOs compared to Aβ monomers in a competitive assay, differentiating ACU193 from other therapeutic monoclonal antibodies, which primarily bind monomer or fibrillar forms of Aβ. Binding of ACU193 to sAβOs is not affected by the presence of a high concentration of Aβ monomers, simulating the environment in the brain (6). These experiments use Aβ-derived diffusible ligands (ADDLs), a synthetic sAβO model system comprising a wide size distribution of toxic sAβOs (9, 10). Evidence for the selectivity of ACU193 for sAβOs over fibrils has been observed with a thioflavin-T binding assay. In this experiment, Aβ aggregation was monitored over time via thioflavin-T, which is known to bind β-sheets within amyloid fibrils. ACU193 immunoreactivity, measured via dot immunoblotting, was greatest when the Aβ population had low thioflavin-T reactivity, thus low fibril content and likely high oligomeric content. As thioflavin-T reactivity, and thus fibril content, increased over time, ACU193 immunoreactivity decreased (data on file). Corroborating this result, ACU193 has little to no binding to thioflavin-S positive amyloid plaque in immunohistochemistry studies (6).
Soluble AβOs interfere acutely with synaptic signaling (10-14), which may be due in part to disruptions in calcium homeostasis as well as inhibition of long-term potentiation (LTP). This aberrant signaling has been associated with inflammatory and neurodegenerative pathologies, such as tau hyperphosphorylation, which contributes to the cognitive impairment of AD (15-18). In rodent hippocampal slice preparations, sAβOs cause rapid inhibition of LTP (10, 14, 19, 20). ACU3B3, the murine IgG1 parent of ACU193, prevented ADDL-associated inhibition of LTP (6, 21).
Increases in intracellular synaptic calcium concentration are associated with neuronal dysfunction and have been implicated in the pathogenesis of AD (22). In primary cultures of transgenic amyloid precursor protein (APP)-PS1 mouse cortical neurons, ADDLs were shown to target synapses in brain tissues, inducing elevated concentrations of neuronal calcium in soma and dendrites. ACU3B3 immuno-depletion prevented this ADDL-elicited calcium elevation, suggesting protection of neuronal calcium homeostasis (6, 23).
In vivo nonclinical studies demonstrate brain penetration and behavioral pharmacologic activity of ACU193/3B3. In vivo administration of ACU193 to Tg2576 mice resulted in a dose-dependent increase of ACU193:sAβO complexes in brain homogenates (data on file). Formation of these ACU193:sAβO complexes in vivo provides direct evidence for ACU193 target engagement in the brain of Tg2576 mice following IV administration. A correlation between cerebrospinal fluid (CSF) levels of sAβOs and cognitive deficits in AD animal models and human patients with AD has been described (24, 25). In vivo activity of ACU3B3, the murine ACU193 parent antibody, has been investigated in three APP transgenic mouse models of AD (i.e., hAPP/SL, Thy1-hAPP/SL, and hAPP/J20) across a battery of eight behavioral tests. Four to 10 weeks of dosing with ACU3B3 in transgenic mice, comprising a broad range of ages, reduced multiple behavioral deficits (i.e., hyperlocomotion, context-dependent habituation, nonspatial learning, and emotionality) (26, 27). Taken together, the results indicate in vivo pharmacodynamic activity of sub-chronic and chronic treatment with ACU3B3 across different ages of APP transgenic animals with variable APP expression levels.
Other monoclonal antibodies that have been or are currently in clinical development for the treatment of AD are not highly selective for sAβOs, albeit with some level of binding variability. Their selectivity profile is predominantly directed to either monomeric or fibrillar Aβ species, due to the later discovery and characterization of sAβO toxicity in AD. Those that bind to amyloid plaque have been associated with higher incidence of ARIA clinically (28). ACU193 is expected to have low risk for ARIA because it has minimal or no binding to amyloid plaques, does not appear to bind to vascular amyloid, and does not increase microhemorrhage in nonclinical studies (6). Given the promising pharmacologic profile of ACU193 and the need to evaluate the “Aβ oligomer hypothesis” in the clinic, ACU193 is now being tested in a phase 1 clinical trial, INTERCEPT-AD.
Traditional phase 1 trials focus solely on safety and pharmacokinetics, which remain primary objectives. Because phase 1 trials for monoclonal antibodies for AD are often conducted in AD patients, they provide the opportunity to begin to assess disease-related outcomes earlier in a drug development program, with the understanding that phase 1 trials are limited by small sample size and short duration. To maximize the scope of information that can be gained from a phase 1 trial in AD patients, the design of the INTERCEPT-AD trial incorporates biomarker measures of target engagement as well as innovative exploratory measures of cognition and cerebral blood flow.
Approach
The clinical trial design for the ACU193 INTERCEPT-AD trial, and overall clinical trial strategy, were based in part on nonclinical data for ACU193, including good laboratory practices toxicology studies. Generally accepted sample sizes for phase 1 studies were considered. Outcomes in INTERCEPT-AD are focused on safety, tolerability, pharmacokinetics, and target engagement. Because ACU193 may improve synaptic function, additional important exploratory measures of potential acute drug effects include assessment of cognition as determined by a computerized cognitive battery and changes in cerebral blood flow as determined by arterial spin labelling (ASL) with magnetic resonance imaging (MRI). An appropriate go/no-go decision to enable a subsequent phase 2/3 study was also considered, as outlined below.
The overall clinical trial strategy, including a planned phase 2/3 trial, was based on the need to rapidly advance ACU193 through clinical trials while maintaining safety. In addition to reviewing nonclinical data for ACU193, trial designs for other AD treatments including other monoclonal antibodies were evaluated (3, 5, 29-31).
Trial Design
As is typical in most early-phase trials testing monoclonal antibodies, patients are utilized in INTERCEPT-AD. This is in part because the drug target, sAβO, is not present in healthy subjects. The patient population being studied in the INTERCEPT-AD trial consists of individuals with early AD, i.e., patients with mild cognitive impairment (MCI) or mild dementia, as intervention in earlier stages of the disease may have a higher probability of benefit. A key inclusion criterion is confirmation of AD with a florbetapir positron emission tomography (PET) scan positive for brain amyloid. APOE genotype is recorded in the study, but treatment groups are not stratified on this variable given the small size of the study. APOE genotyping will be performed because carriers of the APOEε4 allele are at higher risk for ARIA than non-carriers (28). ACU193 is expected to have a low risk of ARIA due to its selectivity for sAβOs and apparent lack of binding to vascular amyloid, but any cases of ARIA will be evaluated for a possible relationship to APOE genotype.
The overall design of the INTERCEPT-AD trial is summarized in Figure 1. Additional information regarding the trial can be found on clinicaltrials.gov (NCT04931459). The study incorporates single ascending dose (SAD) and multiple ascending dose (MAD) cohorts. Doses for SAD cohorts are 2 mg/kg, 10 mg/kg, 25 mg/kg, and 60 mg/kg. SAD cohorts include 8 patients (6:2, active:placebo). As reviewed previously (6), target engagement and behavioral effects were demonstrated in transgenic mice; these studies along with animal toxicology data supported dose selection. Patients in the SAD portion of the study are evaluated in 11 visits ending 140 days after the single dose. Placebo patients from all SAD cohorts are pooled for statistical analyses. For the MAD portion of the study, 3 administrations of ACU193 are given. Dosing intervals were selected using predictions of human exposure based on animal exposure, including measures of serum half-life and CSF pharmacokinetics (data on file). Doses and dosing intervals are 10 mg/kg once every four weeks (Q4W), 60 mg/kg Q4W, and 60 mg/kg once every two weeks (Q2W). MAD cohorts include 10 patients each (8:2, active:placebo). Patients in the MAD portion of the study with Q4W dosing have a total of 7 visits ending 196 days after starting dosing. Patients in the MAD portion of the study with Q2W dosing have a total of 8 visits ending 168 days after starting dosing. Placebo patients from all MAD cohorts are pooled for statistical analyses.
Numerous safety measures are assessed, and patients in the SAD portion of the study are observed as inpatients for 2-4 days. Sentinel dosing is employed for the SAD cohorts, and safety reviews are performed after each cohort has completed dosing prior to opening of the next cohort (see Figure 1 caption for more detail). Possible infusion reactions are carefully assessed, and routine laboratory studies and electrocardiograms are obtained. MRI scans, primarily to assess for the presence of ARIA, are obtained at screening and days 21 and 140 for patients in the SAD cohorts. For patients in the Q4W dosing MAD cohorts, MRIs are obtained at screening and days 28, 70, and 196. For patients in the Q2W MAD cohort, MRIs are obtained at screening and days 28, 70, 168. All MRI centers are qualified by the same external vendor prior to being used for any study assessment. The parameters are optimized to obtain accurate results from different scanner models and manufacturers. The MRI scanner protocol parameters are based on the Alzheimer’s Disease Neuroimaging Initiative (ADNI3) protocol (https://adni.loni.usc.edu/methods/documents/mri-protocols/). Once the protocol parameters are approved a phantom scan is completed and then reviewed by the vendor for qualification.
In addition to extensive safety and tolerability monitoring, pharmacokinetic measures and target engagement are assessed in INTERCEPT-AD. Standard pharmacokinetic parameters are obtained, and comparisons will be made between Q4W and Q2W dosing regimens. Target engagement in the central compartment will be assessed by measuring concentrations of ACU193 bound to sAβOs in CSF.
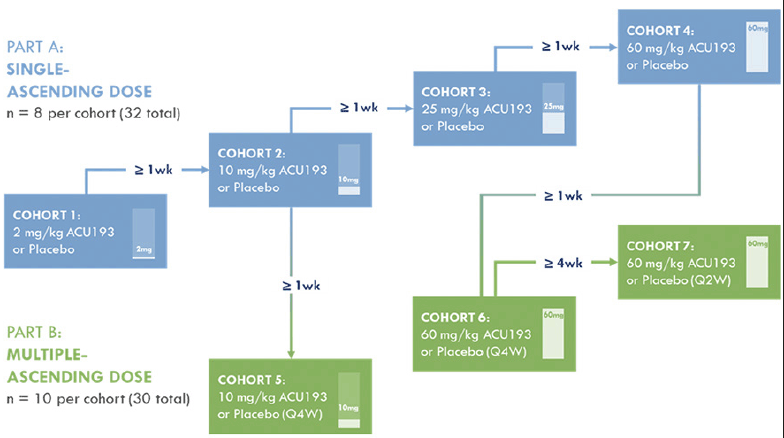
Figure 1. Trial schematic of INTERCEPT-AD
For Part A (SAD): a sentinel dosing scheme is followed within each cohort, whereby the third participant is dosed at a minimum of 48 hours after the first two participants are dosed (one receives blinded ACU193 and the other blinded placebo) and safety data are reviewed. Dosing of Cohorts 1-4 begins at least one week after all participants in the immediately preceding lower-dose cohort receives one administration of study drug (ACU193 or placebo) and safety data are reviewed by a blinded internal safety team; pharmacokinetic data are also reviewed periodically. If necessary, an independent, unblinded data monitoring committee (DMC) may also review these data and advise on dose escalation. For Part B (MAD): Dosing of Cohort 5 begins at least one week after all participants in Cohort 2 receive one administration of study drug (ACU193 [10 mg/kg] or placebo) and safety data are reviewed. Dosing of Cohort 6 begins at least one week after all participants in Cohort 4 receive one administration of study drug (ACU193 [60 mg/kg] or placebo) and safety data are reviewed. Dosing of Cohort 7 begins after four or more participants in Cohort 6 are administered two doses of study drug (ACU193 [60 mg/kg] or placebo Q4W) and the Cohort 6 safety data are reviewed by the blinded internal safety team. PK data are also reviewed periodically. If necessary, an independent, unblinded DMC may also review the data and advise on dose escalation. Q2W = once every two weeks; Q4W = once every four weeks
While changes in cognitive measures are not expected in INTERCEPT-AD due to the short treatment duration and small sample size, standard measures of cognition and function are obtained during the trial including the Alzheimer’s Disease Assessment Scale – cognition (ADAS-cog) and the Clinical Dementia Rating (CDR) scale. Additionally, a battery of brief computerized cognitive tests, designed to provide a more sensitive assessment of a breadth of cognitive domains relevant to early AD, are obtained at multiple timepoints in the trial. Assessment and scoring are fully automated, and the battery is supervised by a trained administrator. The individual tests included in the battery, which can be administered in approximately 30 minutes, are listed in Table 1. For the SAD cohorts, the battery is administered at baseline and days 2, 3, 4, 7, and 21. For MAD cohorts using Q4W dosing, the battery is administered at baseline and days 7, 28, 56, 70, and 196. For the MAD cohort using Q2W dosing, the battery is administered at baseline and days 7, 28, 42, and 168.
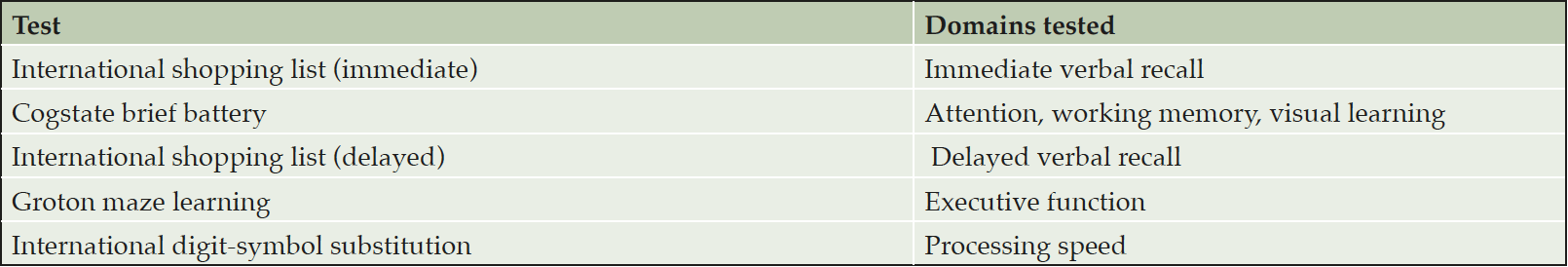
Table 1. Computerized cognitive test battery
Reduced blood flow (hypoperfusion) to brain tissue precedes atrophy in AD (32), and co-localizes with hypometabolism measured by PET (33), suggesting an association with AD-related neuropathology. Thus, changes in cerebral blood flow may be an early biomarker of AD. Therefore, while exploratory in this phase 1 trial, cerebral blood flow is assessed by an ASL pulse sequence as part of the MRI. ASL images are acquired via 3D pseudo-continuous ASL or 3D pulsed ASL sequences (34), depending on availability, and according to sequence parameter guidelines of the longitudinal observational study ADNI3 (https://adni.loni.usc.edu/adni-3/). Images are collected as part of the MRI assessment at screening, day 21, and end-of-study for the SAD cohorts and at screening, days 28, 70, and end-of-study for the MAD cohorts.
At the time of this writing, patients have entered SAD and MAD cohorts and the trial is ongoing.
The overall clinical strategy for ACU193 includes two key decision-making milestones. The go-no/go decision to move from phase 1 to phase 2/3 will based on the following: safety and tolerability at doses that are projected to have pharmacological activity based on nonclinical data; pharmacokinetic parameters that are appropriate for continued development; and target engagement at doses that have acceptable safety and tolerability. The second milestone is based on an interim analysis in the phase 2/3 study. The phase 2/3 study will be initiated with sample sizes typical of a phase 2 study; however, an interim analysis will enable a go/no-go decision to increase the sample size for full powering of a phase 3 trial. The interim analysis will be based on key clinical measures, but also various biomarkers, which could include blood and CSF p-tau, blood and CSF neurofilament light (NfL), computerized cognitive testing, and cerebral blood flow changes measured by ASL MRI, that may be integrated to support this decision making (35). The importance of go/no-go decision making in drug development has been recently reviewed (35). Assuming a positive interim assessment and pending discussions with regulators, this phase 2/3 study could serve as a registration trial.
Discussion
While promising results from Aβ-targeting antibodies have been recently published (2, 3, 5), Alzheimer’s disease remains a large and growing public health problem that can only be addressed with treatments that result in clinically meaningful disease modification accompanied by acceptable safety and tolerability. While the “amyloid hypothesis” has in the past been viewed by some as a monolithic entity, clinical trial data are now showing that more specific targeting of various Aβ or amyloid species is likely to be instructive. The monomeric Aβ peptide is highly aggregate-prone, able to form soluble oligomers, soluble or insoluble protofibrils, and insoluble fibrils or amyloid plaque (1). Statistically significant differences or trends favoring treatment have been shown in clinical outcomes for antibodies targeting amyloid plaques [aducanumab (2) and donanemab (3)], soluble protofibrils [lecanemab (5)] and Aβ monomers [solanezumab (36)]. Despite these similarities, substantial differences in rates of ARIA-E are seen, with plaque-targeting antibodies causing higher incidence of ARIA-E (37) than those that target other forms of Aβ. The incidence of ARIA with an antibody that selectively targets sAβOs is not yet known but is expected to be lower compared to that of plaque-targeting antibodies.
Progress in the understanding of underlying disease pathology and the use of trial designs incorporating biomarkers as well as clinical outcome measures can accelerate the development of AD therapeutics. The ACU193 INTERCEPT-AD trial utilizes a study design that incorporates novel measures in AD patients, allowing assessment of disease-related outcomes earlier in a clinical development program. The computerized cognitive assessments used in the trial have been validated in multiple clinical trials, are intended to address cognitive domains under-represented by traditional tools, and have previously shown sensitivity in proof-of-concept studies in AD, including the early and more sensitive detection of drug effect (38-41). Because of the brevity of the computerized cognitive test battery, it can be administered multiple times, thus reducing variability and potentially allowing detection of a drug effect that may not be evident using more standard cognitive scales, such as the Alzheimer’s Disease Assessment Scale – cognition (ADAS-cog). Many studies have shown a decrease in cerebral blood flow in AD, and more recently, changes in cerebral blood flow as assessed by ASL have been shown to correlate with tau-PET in the entorhinal cortex in AD (42). Reduced cerebral blood flow may be an early biomarker of AD and can be measured as part of the safety MRIs that are already included in a phase 1 AD study. Because of the dynamic nature of this measure, it may have the capability to detect potential acute drug effects during the short duration of treatment in a phase 1 trial.
The go/no-go decision in the INTERCEPT-AD trial will be based on safety and tolerability, pharmacokinetics, and evidence of target engagement. The use of digital and imaging biomarkers in INTERCEPT-AD, i.e., computerized cognitive testing and regional cerebral blood flow changes from ASL MRI, will provide additional data, which, while exploratory, would strengthen a decision to move to an adaptive phase 2/3 study.
For the adaptive phase 2/3 study, a go/no-go decision to increase the size of the trial to a phase 3 registration study will be made based on an interim analysis of phase 2 data. Other clinical trials in neurodegenerative disease have used adaptive designs to identify effective doses more quickly or to avoid additional use of resources if a trial has a low probability of success (43, 44). It is also important to acknowledge that there are risks associated with an interim analysis approach to expand to a phase 3 study, as meeting criteria for expansion does not guarantee a successful conclusion of the trial. The fact that the interim analysis will not be used to determine futility is important to note; if the trial is not expanded to a phase 3 study, it will be completed as a phase 2 study.
Taken together, the ACU193 clinical development plan incorporates several advances in AD research. The net result of these improvements is that the development plan, using biomarkers and adaptive design, moves from a first-in-human trial directly to a registration-quality phase 2/3 trial rather than the traditional phase 1, phase 2, and phase 3 sequence. Safety can be maintained with the active use of a data monitoring committee (DMC) during the trials. Go/no-go decision making can be enhanced with the use of imaging, biochemical, and digital biomarkers. In addition to the use of novel trial designs, this development plan builds on previous research showing encouraging drug effects in patients with early AD, a patient population that has recently become widely recognized and utilized. Finally, the “Aβ oligomer hypothesis” is now being tested in the clinic with ACU193. ACU193 provides an opportunity to assess whether selectively targeting toxic sAβOs results in meaningful efficacy and lower risk of ARIA as compared to other monoclonal antibody treatments. The use of new scientific information and novel trial designs can more rapidly assess the oligomer hypothesis and can more broadly accelerate the development of more effective treatments for patients with AD.
Funding: Acumen Pharmaceuticals, Inc. is the sponsor of this study and provided support for the preparation of this manuscript.
Acknowledgements: The efforts of patients and study partners in the INTERCEPT-AD and many other clinical trials are greatly appreciated. The involvement of clinical trial sites which can participate in studies that utilize novel study designs is also greatly appreciated.
Ethical standards: Institutional Review Boards (IRBs) approved the INTERCEPT-AD study, and all participants gave informed consent before participating.
Potential conflicts of interest: Eric Siemers, Janice Hitchcock, Karen Sundell, Robert Dean, Jasna Jerecic, Erika Cline, Kent Iverson, Jerome Moore, and Russell Barton are employees or consultants for Acumen Pharmaceuticals, Inc. Chris Edgar is an employee of Cogstate Ltd. Richard Manber, Niccolo Fuin, and Tanya Poppe are employees of Ixico Plc. © The Authors 2022
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. Siemers E, Aisen PS, Carrillo MC. The Ups and Downs of Amyloid in Alzheimer’s. J Prev Alzheimers Dis. 2022;9(1):92-95. doi:10.14283/jpad.2021.54
2. Budd Haeberlein S, Aisen PS, Barkhof F, et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. The Journal of Prevention of Alzheimer’s Disease. 2022. doi:10.14283/jpad.2022.30
3. Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in Early Alzheimer’s Disease. New England Journal of Medicine. 2021. doi:10.1056/NEJMoa2100708
4. Logovinsky V, Satlin A, Lai R, et al. Safety and tolerability of BAN2401–a clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimers Res Ther. 2016;8(1):14. doi:10.1186/s13195-016-0181-2
5. Swanson CJ, Zhang Y, Dhadda S, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimer’s Research & Therapy. 2021;13(1). doi:10.1186/s13195-021-00813-8
6. Krafft GA, Jerecic J, Siemers E, Cline EN. ACU193: An Immunotherapeutic Poised to Test the Amyloid β Oligomer Hypothesis of Alzheimer’s Disease. Frontiers in Neuroscience. 2022;16. doi:10.3389/fnins.2022.848215
7. Goure WF, Krafft GA, Jerecic J, Hefti F. Targeting the proper amyloid-beta neuronal toxins: a path forward for Alzheimer’s disease immunotherapeutics. Alzheimer’s Research & Therapy. 2014;6(4):42. doi:10.1186/alzrt272
8. Savage MJ, Kalinina J, Wolfe A, et al. A Sensitive Aβ Oligomer Assay Discriminates Alzheimer’s and Aged Control Cerebrospinal Fluid. Journal of Neuroscience. 2014;34(8):2884-2897. doi:10.1523/jneurosci.1675-13.2014
9. Chromy BA, Nowak RJ, Lambert MP, et al. Self-assembly of Abeta(1-42) into globular neurotoxins. Biochemistry. 2003;42(44):12749-60. doi:10.1021/bi030029q
10. Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95(11):6448-53. doi:10.1073/pnas.95.11.6448
11. Klyubin I, Betts V, Welzel AT, et al. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28(16):4231-7. doi:10.1523/JNEUROSCI.5161-07.2008
12. Shankar GM, Li S, Mehta TH, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14(8):837-42. doi:10.1038/nm1782
13. Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572(Pt 2):477-92. doi:10.1113/jphysiol.2005.103754
14. Walsh DM, Klyubin I, Shankar GM, et al. The role of cell-derived oligomers of Aβ in Alzheimer’s disease and avenues for therapeutic intervention. Biochem Soc Trans. 2005;33(Pt 5):1087-90. doi:10.1042/BST20051087
15. Cleary JP, Walsh DM, Hofmeister JJ, et al. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8(1):79-84. doi:10.1038/nn1372
16. Poling A, Morgan-Paisley K, Panos JJ, et al. Oligomers of the amyloid-beta protein disrupt working memory: confirmation with two behavioral procedures. Behav Brain Res. 2008;193(2):230-4. doi:10.1016/j.bbr.2008.06.001
17. Reed MN, Hofmeister JJ, Jungbauer L, et al. Cognitive effects of cell-derived and synthetically derived Aβ oligomers. Neurobiol Aging. 2011;32(10):1784-94. doi:10.1016/j.neurobiolaging.2009.11.007
18. Townsend M, Cleary JP, Mehta T, et al. Orally available compound prevents deficits in memory caused by the Alzheimer amyloid-beta oligomers. Ann Neurol. 2006;60(6):668-76. doi:10.1002/ana.21051
19. Barghorn S, Nimmrich V, Striebinger A, et al. Globular amyloid beta-peptide oligomer – a homogenous and stable neuropathological protein in Alzheimer’s disease. J Neurochem. 2005;95(3):834-47. doi:10.1111/j.1471-4159.2005.03407.x
20. Rammes G, Hasenjager A, Sroka-Saidi K, Deussing JM, Parsons CG. Therapeutic significance of NR2B-containing NMDA receptors and mGluR5 metabotropic glutamate receptors in mediating the synaptotoxic effects of beta-amyloid oligomers on long-term potentiation (LTP) in murine hippocampal slices. Neuropharmacology. 2011;60(6):982-90. doi:10.1016/j.neuropharm.2011.01.051
21. Cline, E., Viola, K., Klein, W., Wang, X., Bacskai, B., Rammes, G., Dodart, J., Palop, J., Siemers, E., Jerecic, J., Krafft, G. Synaptic intervention in Alzheimer’s disease: soluble Aβ oligomer directed ACU193 monoclonal antibody therapeutic for treatment of early Alzheimer’s disease. J Prevent Alzheimer’s Dis. 2019;6(S1):S151.
22. Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30(36):11938-50. doi:10.1523/JNEUROSCI.2357-10.2010
23. Wang X, Kastanenka KV, Arbel-Ornath M, et al. An acute functional screen identifies an effective antibody targeting amyloid-β oligomers based on calcium imaging. Scientific Reports. 2018;8(1):4634. doi:10.1038/s41598-018-22979-2
24. Goure WF, Krafft GA, Jerecic J, Hefti F. Targeting the proper amyloid-beta neuronal toxins: a path forward for Alzheimer’s disease immunotherapeutics. Alzheimers Res Ther. 2014;6(4):42. doi:10.1186/alzrt272
25. Savage MJ, Kalinina J, Wolfe A, et al. A sensitive Aβ oligomer assay discriminates Alzheimer’s and aged control cerebrospinal fluid. J Neurosci. 2014;34(8):2884-97. doi:10.1523/JNEUROSCI.1675-13.2014
26. Dodart J-C, Nay, S., Monbureau, M., Hefti, F. F., Jerecic, J., Shamloo, M. Passive immunization with the anti-Aβ oligomer antibody ACU-3B3 improves behavioral deficits in hAPPSL transgenic mice. Presented at: Society for Neuroscience; 2014; Session 135.04/G1.
27. Ma K, Martinez-Losa, M.M., Xue, Y., Khan, A., Ho, K., Dodart, J.-C., Jerecic, J., Cobos, I., Palop, J.J. . P2-063: Soluble Aβ-Oligomer–Selective Antibody ACU3B3 Reduces Amyloid Pathology & Improves Multiple Behavioral Domains in a Mouse Model of AD. Alzheimer’s and Dementia. 2019;15(7S Part 11):595. doi:10.1016/j.jalz.2019.06.2470
28. Withington CG, Turner RS. Amyloid-Related Imaging Abnormalities With Anti-amyloid Antibodies for the Treatment of Dementia Due to Alzheimer’s Disease. Front Neurol. 2022;13:862369. doi:10.3389/fneur.2022.862369
29. Doody RS, Raman R, Farlow M, et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med. 2013;369(4):341-50. doi:10.1056/NEJMoa1210951
30. Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):311-21. doi:10.1056/NEJMoa1312889
31. Ostrowitzki S, Lasser RA, Dorflinger E, et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther. 2017;9(1):95. doi:10.1186/s13195-017-0318-y
32. Jack CR, Jr., Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9(1):119-28. doi:10.1016/S1474-4422(09)70299-6
33. Chen Y, Wolk DA, Reddin JS, et al. Voxel-level comparison of arterial spin-labeled perfusion MRI and FDG-PET in Alzheimer disease. Neurology. 2011;77(22):1977-85. doi:10.1212/WNL.0b013e31823a0ef7
34. Alsop DC, Detre JA, Golay X, et al. Recommended implementation of arterial spin-labeled perfusion MRI for clinical applications: A consensus of the ISMRM perfusion study group and the European consortium for ASL in dementia. Magn Reson Med. 2015;73(1):102-16. doi:10.1002/mrm.25197
35. Wessels AM, Edgar CJ, Nathan PJ, Siemers ER, Maruff P, Harrison J. Cognitive Go/No-Go decision-making criteria in Alzheimer’s disease drug development. Drug Discov Today. 2021;26(5):1330-1336. doi:10.1016/j.drudis.2021.01.012
36. Honig LS, Vellas B, Woodward M, et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N Engl J Med. 2018;378(4):321-330. doi:10.1056/NEJMoa1705971
37. Barakos J, Purcell D, Suhy J, et al. Detection and Management of Amyloid-Related Imaging Abnormalities in Patients with Alzheimer’s Disease Treated with Anti-Amyloid Beta Therapy. J Prev Alzheimers Dis. 2022;9(2):211-220. doi:10.14283/jpad.2022.21
38. Grove RA, Harrington CM, Mahler A, et al. A randomized, double-blind, placebo-controlled, 16-week study of the H3 receptor antagonist, GSK239512 as a monotherapy in subjects with mild-to-moderate Alzheimer’s disease. Curr Alzheimer Res. 2014;11(1):47-58. doi:10.2174/1567205010666131212110148
39. Karin A. Validation of Cogstate cognitive test battery used in a clinical study with Alzheimer’s disease patients. Master’s Thesis. 2012.
40. Maher-Edwards G, De’Ath J, Barnett C, Lavrov A, Lockhart A. A 24-week study to evaluate the effect of rilapladib on cognition and cerebrospinal fluid biomarkers of Alzheimer’s disease. Alzheimers Dement (N Y). 2015;1(2):131-140. doi:10.1016/j.trci.2015.06.003
41. Scheltens P, Hallikainen M, Grimmer T, et al. Safety, tolerability and efficacy of the glutaminyl cyclase inhibitor PQ912 in Alzheimer’s disease: results of a randomized, double-blind, placebo-controlled phase 2a study. Alzheimers Res Ther. 2018;10(1):107. doi:10.1186/s13195-018-0431-6
42. Rubinski A, Tosun D, Franzmeier N, et al. Lower cerebral perfusion is associated with tau-PET in the entorhinal cortex across the Alzheimer’s continuum. Neurobiology of Aging. 2021. doi:10.1016/j.neurobiolaging.2021.02.003
43. Lang AE, Siderowf AD, Macklin EA, et al. Trial of Cinpanemab in Early Parkinson’s Disease. N Engl J Med. 2022;387(5):408-420. doi:10.1056/NEJMoa2203395
44. Satlin A, Wang J, Logovinsky V, et al. Design of a Bayesian adaptive phase 2 proof-of-concept trial for BAN2401, a putative disease-modifying monoclonal antibody for the treatment of Alzheimer’s disease. Alzheimer’s & Dementia: Translational Research & Clinical Interventions. 2016;2(1):1-12. doi:10.1016/j.trci.2016.01.001