
M. Tatar
Department of Ecology, Evolution and Organismal Biology
Corresponding Author: Marc Tatar, Department of Ecology, Evolution and Organismal Biology, Box GW, Walter Hall Brown University, Providence RI 02912, USA, Office: +1 401-863-3455, Fax: +1 401-863-2166, Marc_Tatar@Brown.edu
J Prev Alz Dis 2022;4(9):580-588
Published online August 31, 2022, http://dx.doi.org/10.14283/jpad.2022.68
Abstract
Alzheimer’s Disease is a progressive manifestation of aging associated with accumulated Amyloid β. It remains frustratingly unclear why this protein accumulates and how it contributes to Alzheimer’s Disease pathology. In one recent hypothesis, Amyloid β is suggested to function as an antimicrobial peptide in innate immune defense within the brain, where Amyloid β gains toxicity when it becomes abundant. This essay proposes an evolutionary explanation for why Amyloid β expression is regulated at an optimum based on its function as a defense and how this leads to disease. Among its potential physiological functions, Amyloid β confers benefits to reduce direct pathogen damage while this simultaneously entails cellular cost of defense. Optimal Amyloid β expression occurs when the gain in fitness from an incremental increase is balanced by the marginal cost of this increase. It proposes that natural selection acting upon the young favored systems to maintain Amyloid β at an optimal level through mechanisms that induce the defense and repress its expression. With age, the force of natural selection declines and permits mechanisms of negative feedback repression to degenerate. Consequently, Amyloid β is expressed beyond its optimum. Age also elevates cumulative pathogen exposure, reduces pathogen barriers and reactivates latent pathogens. The net effect is elevated, chronic induction of Amyloid β in the brain. The model recommends attention to innate immune negative regulation in the brain to discover ways to restore these functions toward a youthful state in the elderly.
Key words: Alzheimer’s disease, Amyloid beta, signal detection, antimicrobial protection hypothesis, defense regulation.
Alzheimer’s Disease (AD) is a prevalent form of age-associated neurodegeneration. The incidence of AD accelerates each decade after age 65 years, while genetic factors predispose individuals to higher age-associated risk and to early-onset familial Alzheimer’s Disease (1). Amyloid beta protein (Aβ), a recognized contributing factor to this pathology (2, 3), is a cleavage product of amyloid precursor protein (APP). Aβ is generated within the Golgi apparatus and endoplasmic reticulum through sequential cleavage of APP by BACE1 and γ-secretase. γ-secretase primarily produces the Aβ1-40 isoform and secondarily the longer Aβ1-42 species. Secreted from cells, these Aβ occur as monomers, low-molecular weight oligomers, protofibrils and mature β-sheet fibrils (amyloid plaques). Aβ1-42 has greater propensity to form oligomers and higher order β-amyloid sheets (4). Given its phylogenetic prevalence and its distribution in many tissues, Aβ is likely to have positive functions (5-9). This essay proposes AD arises from the way natural selection has acted in the young to optimize the expression of Aβ based on its innate immune function relative to the costs of this defense. The concept helps bridge explanations between the mechanisms that cause AD (the ‘how’ of Tinbergen, (10)) and why evolution has produced this trait. ‘Making sense’ of this evolution (11) may suggest therapeutic approaches where the essential role of feedback is recognized to maintain optimized Aβ expression.
The negative consequences of Aβ are demonstrated when production is experimentally studied in mice. Animals expressing human forms of APP and a mutant presenilin γ-secretase proteolytic subunit have β-amyloid plaques, neuroinflammation and impaired cognition, although without tau neurofibrillary tangles (12-14). Neurofibrillary tangles are observed in human stem cell-derived neurons with familial Alzheimer’s disease mutations (15). These and related data support a version of the amyloid hypothesis whereby age-dependent increase in Aβ induces tauopathy that leads to neurological decay (16). Nonetheless, it is unclear whether accumulated Aβ is sufficient to cause AD (17, 18). Recent clinical trials with monoclonal antibodies reduce the level of Aβ but have little effect on dementia (19, 20). Furthermore, some elderly individuals have high cognitive function despite widespread Aβ and NFT (21, 22). These outcomes may arise in part because Aβ has unrecognized properties. In particular, Robinson and colleagues proposed Aβ has physiological roles including to protect from infection, to repair the blood brain barrier, and to regulate synapses (5, 6). Emphasizing innate immunity, Moir, Tanzi and colleagues demonstrated Aβ can function as an antimicrobial peptide, positing the antimicrobial defense hypothesis of AD (7-9). Antimicrobial peptides are small peptides with amphipathic secondary structure produced by cells to defeat bacterial, fungal, viral and protozoan infections (23). Thus, Aβ may have evolved as a useful but dangerous defense system. From this perspective, the prevention of AD might be framed by asking what are the mechanisms that maintain the optimal level of Aβ activity and how do these mechanisms fail during aging? Understanding why Aβ has an optimal level of expression will suggest how cells maintain this level and what features may therefore degenerate as we age. I propose that Aβ expression is tuned in the young as an evolutionary compromise between its benefits and costs, and this optimal level is maintained by mechanisms of induction and negative regulation. When the force of natural selection weakens at advanced age, there is less selection to prevent degeneration of the negative regulatory system. Infection, viral reactivation or even ‘sterile’ agents may then stimulate Aβ beyond its optimal level. Aβ is always dangerous, even at young ages, but there is little fitness cost to its over-production at advanced age and we experience neurodegeneration. From this view, a potential strategy to maintain neuronal health in aging might seek ways to retain the negative regulatory network of Aβ at a youthful state.
Aβ as an optimized innate immune defense
Defense system evolution
Randolph Nesse applied signal detection theory to understand the evolution of defense systems (24). Fever, pain, fear, nausea, and immunity are defenses that protect individuals from acute injury, threats and infection. Such defenses are activated only when needed because they incur costs alongside their benefits. Nesse proposed defense activity is optimized to maximize fitness based on the relationship between their costs and benefits. Fitness is the relative contribution to subsequent generations made by an individual or genotype. Thus, damage caused by a pathogen – its direct virulence – reduces host fitness. Host immunity mitigates the direct virulence by inhibiting the growth or action of the pathogen. Graphically, the direct virulence (DV) function describes how the fitness costs from pathogen damage is reduced by the expressed level of host immune defense (Figure 1A, adapted from Nesse (24)). The shape of this function depends on specific details of the pathogen and the efficiency of the defense, but in general it will reflect diminishing returns.
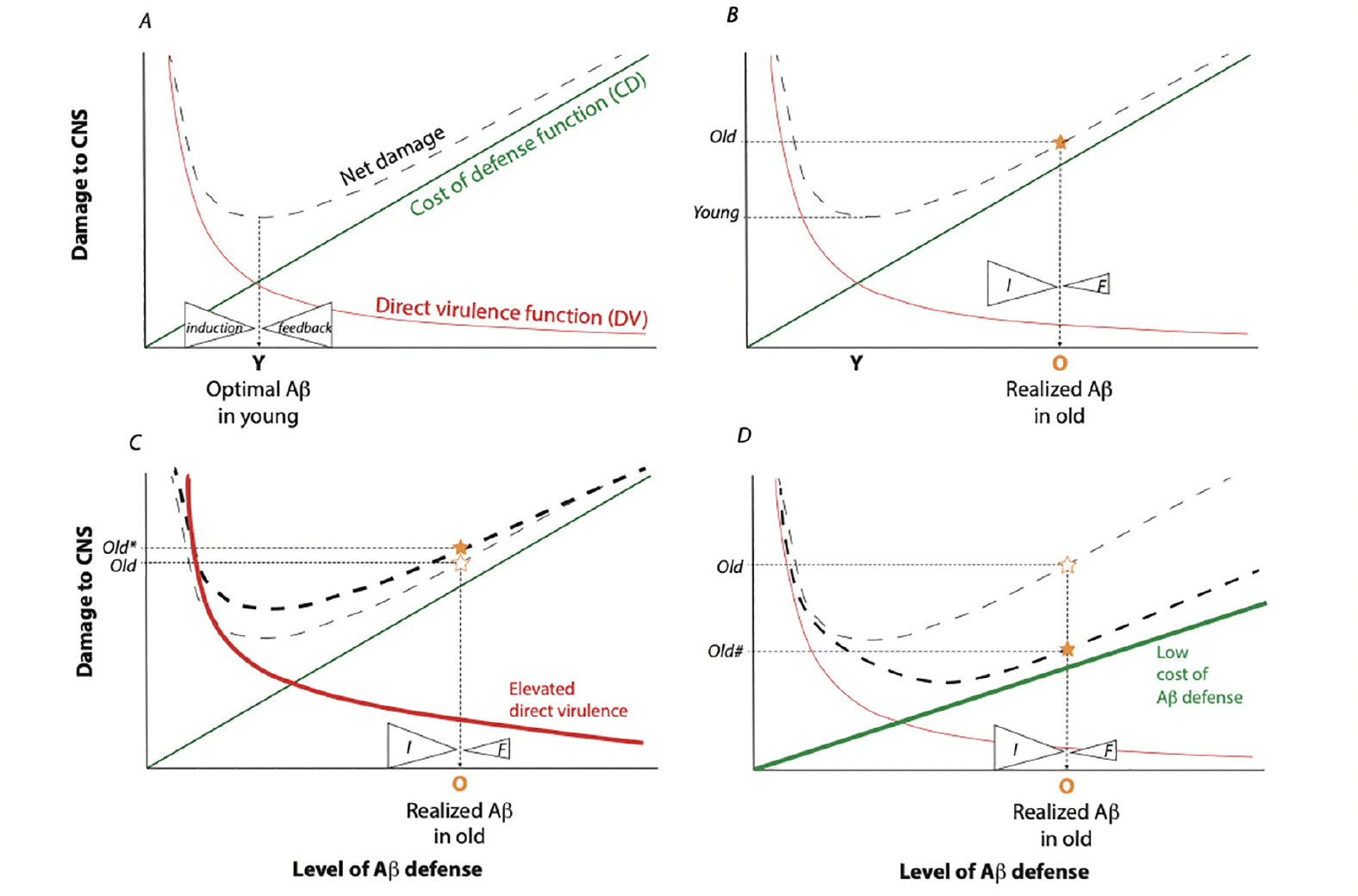
Figure 1. Model for the evolution of neurodegeneration as an outcome of defense optimization
Model built from the signal detection theory of Nesse (24). Each figure represents CNS net damage as a function of induced Aβ. Infection by bacteria, fungi, or viruses (or activated retrotransposons) produces direct damage to the CNS, represented as the Direct Virulence Function (DV), red lines. The DV is decreased in response to induced innate immune defense, here specifically by the level of Aβ. Expression of Aβ entails costs (damage) within the CNS, represented by the Cost of Defense function (CD) green lines. The sum of DV and CD yields the Net Damage (dashed lines). Fitness is maximized by strong selection acting in the young, which minimizes net damage. A) This model; proposes the optimal level of expressed Aβ in the young (Y) is maintained by counterbalanced mechanisms of Aβ induction (I) and negative feedback (F). B) In the aged, the force of natural selection is weak, permitting feedback mechanisms to degenerate such that ineffective feedback is less able to counterbalance the force of Aβ induction. Realized Aβ expression increases (O) and elevates the level of net CNS damage (star). This damage is manifest as neurodegeneration. C) Induction may also increase with age as old individuals acquire infection, experience reactivation of latent pathogens and loose pathogen barriers. This will elevate the DV function (heavy red line) and further increase the CNS net damage (Old*). D) Some individuals may have a low Aβ defense cost function (heavy green line). With lost capacity for negative feedback during aging, they still accumulate Aβ but the CNS net damage function is reduced (Old#). These old individuals would not present with Aβ-associated dementia yet their brains may have accumulated Aβ.
Defense mechanisms are expensive. Immune defense mechanisms incur fitness costs because they require physiological resources, and can also directly damage cells and disrupt tissue function (25, 26). As an example, pulmonary damage upon viral infection in humans includes effects from host immune-mediated responses that disrupt tissue architecture (27). In Drosophila, activated NF-κB induces an antimicrobial response but strongly represses fecundity, even when the activating stimulus is sterile (28). In mammals, TNFα induces a protective inflammatory response through degranulation yet this impairs vascular hemostasis (29). These costs are graded functions when they increase with the level of defense (Fig 1, cost of defense (CD) function). Nesse assumed a linear cost of defense although these functions may accelerate as occurs when fever produces hyperpyrexia or when highly activated macrophages produce systemic inflammatory disorders (30).
The shapes of the DV and CD functions specify the optimal defense activity. This point may sit where the level of defense minimizes the total cost, that is where the DV and CD intersect although Nesse emphasizes the optimum occurs where the marginal gain from additional defense is no longer positive. The optimum is determined by the relative rate of change between the DV and the CD, and it may be greater than expected for instance when DV declines quickly relative to the cost for increased defense (Fig 1A).
The aim is to understand how selection has shaped mechanisms to maintain defenses at their optimum in the context of this model. That is, what proximal mechanistic systems exist that satisfy the forces impacting defense evolution? I propose the optimum level of defense is controlled by counter-balancing induction and negative feedback, and this can maintain a quasi-homeostatic level of defense until an infection is cleared. The defense may be terminated in a gradual manner if the force of negative regulation decreases continuously when the inductive signals are reduced, as described for how inflammation is resolved by Dunster et al (31). If the negative regulation persists somewhat after inductive signals wane, the system may exhibit hysteresis whereby the defense abruptly terminates (32). Negative feedback, the essential ‘how’ in this model, occurs in many defense systems (29, 33). For instance, neutrophils produce arachidonate lipids that convert into proinflammatory leukotriene B4 at the site of injury. Arachidonate is subsequently converted by the affected tissue into lipoxins that block neutrophil intrusion into the tissue (34). In a second case, the A20 zinc finger domain protein is induced by TNF and TLR signaling via NF-κB where A20 subsequently ubiquitinates signaling elements of the NF-κB pathway to dampen inflammation (35). As a final example, experimental encephalomyelitis stimulates immunoglobulin OX2 within macrophages, which slows macrophage tissue influx and limits autoimmune damage (36). Maintaining an optimal defense level requires more than losing the inductive stimuli — it likely involves active systems of negative regulation. Thus, understanding the balance between the inductive and negative feedback systems of Aβ as a defense is an essential tool toward therapeutic management of AD.
Infection and immune defense of the brain
The evolution of defense predisposes humans to neurodegeneration because the brain is exposed to pathogens (37) and the immune response of the brain provides protection with associated costs. Many defensive immune responses may contribute to neurodegeneration (38-40), of which amyloid as an antimicrobial peptide is one emerging hypothesized component. Here I consider this innate immune mechanism of Aβ as a model for how defenses are turned on and turned off. The principle is general to all defense systems of the brain and can inform new ways to manage neurodegeneration in aging.
The CNS copes with viral infections as well as bacterial and fungal pathogens. These can induce amyloid and associate with AD (41, 42). Herpes simplex virus-1 (HSV-1) is a frequently latent infection within the peripheral nervous system (43). Recent work with mice and cell culture show reactivation of herpes in the elderly can induce CNS amyloid pathology (44, 45). The neurotrophic protozoan Toxoplasma gondii is likewise argued to associate with AD etiology (46). Long-term infection by T. gondii in humans is widespread and largely benign except in immune compromised individuals (47, 48). Based on meta-analysis, the risk of AD is about 50% greater among T. gondii seropositive individuals (49). Human cytomegalovirus (HCMV) is also a common, latent pathogen, although data do not consistently associate it with AD (50, 51). Ancestral retrotransposable elements within our genomes such as LINE-1 can be activated with age in several tissues (52, 53). In cultured cells these LINE-1 elements may be perceived by the Type I interferon response system and thus induce an inflammatory response (53). Aside from viruses, subclinical fungal and bacterial infections occur in the human brain and associate with AD (54, 55). Candida protein and DNA has been detected in the brains of AD patients, while Candida glabrata, C. albacans, Penicillium notatum and Syncephalastrum racemosum were seen in neurons of AD but not control brains (56). Whether these fungal infections cause AD or are secondary consequences of neurodegeneration remains unknown. Chlamydia pneumoniae is a ubiquitous, intracellular respiratory bacterial parasite. Initial reports found C. pneumoniae to associate with AD, while intranasal infection with C. pneumoniae activated astrocytes and produced Aβ deposits in the brain of wildtype mice (42, 57). Collectively these examples show that various microbes that associate with AD are available to stimulate defense systems in the CNS.
Upon new infection or pathogen reactivation, the brain is defended by the innate immune system. Microglia are innate immune sentinels of the CNS, coordinating phagocytosis and producing inflammatory cytokines (58-60). Glial astrocytes directly contribute to the innate immune inflammatory response and are likely the key source of amyloid production (61, 62). Neurons themselves generate Aβ when APP is processed within mitochondria-associated endoplasmic reticulum membranes (63). In these circumstances it is proposed with some debate (64) that Aβ acts in a defensive role where it provides antimicrobial activity (9). Synthetic Aβ kills Streptococcus and E. coli in vitro (7), while synthetic Aβ protects cultured cells from virus (65, 66). C. elegans engineered to express human Aβ42 are protected from Candida albicans infection, while 5XFAD mice resist cerebral Salmonella infection (8). Several mechanisms of protection by Aβ are proposed. Aβ oligomers bind microbe cell wall carbohydrates, cause agglutination and inhibit microbe adhesion to host cells (8). High concentrations Aβ porate microbes as do other AMPs (67). Aβ plaques may entrap viruses and bacteria, activating microglia migration (68-70). These studies suggest Aβ potentially acts within the brain as a defense to reduce the direct harm from pathogens.
Mechanisms are also proposed for how Aβ incurs costs (71). The oligomers of Aβ are considered to be toxic (72). Soluble Aβ oligomers may disrupt synaptic receptors (73). Aβ may also produce ion channels in liposomes and thereby disrupt cellular ionic balance (74, 75). Zaretsky and Zaretskaia (76) propose extracellular Aβ40 and Aβ42 enter cells by endocytosis into lysosomes where protein degradation produces Aβ fragments that porate the lysosome, the plasma membrane and mitochondria. As lysosomes are permeabilized, autophagy and mitochondrial function are disrupted, producing hallmarks of AD pathology (77, 78). Aβ oligomers may also cross-seed intracellular tau through the action of a common epitope, contributing to tau neurofibrillary tangles (79). There are many potential ways Aβ can be toxic, and these incur costs when Aβ is induced as a defense.
The balance of Aβ
The costs and benefits of neuroprotective defense will be balanced by natural selection, where the impact of the pathogen on fitness is the sum of direct harm from the microbe plus damage caused by the induced defense. Aβ incurs defense costs when it acts as an induced antimicrobial peptide to limit microbe damage but the net CNS damage is less than would occur without the defense. I propose natural selection molds mechanisms of Aβ induction and negative feedback to achieve an optimal level of Aβ activity. If feedback systems involve hysteresis, this will generate a bistable equilibrium that abruptly activates Aβ to its optimum then abruptly terminates the defense once the infection is cleared. Appreciating these proximal requirements, many studies identify mechanisms for how Aβ is induced while others identify potential systems for negative feedback to stop production and clear Aβ.
Aβ is synthesized from the transmembrane amyloid precursor protein APP, which is processed by two related pathways (80). APP is made into non-amyloid P3 peptide via cleavage by α-secretase followed by γ-secretase. Alternatively, in the amyloidogenic pathway APP is cleaved by β-secretase (BACE1) and γ-secretase to release Aβ peptides of 39 to 42 amino acids along with the APP intracellular domain protein. Points of control occur in APP processing when BACE1 activity is rate-limiting, although regulation of this step is not well understood (81). The transcription of β-secretase is induced by many stressors, including HSV infection (82). The enzymatic activity of BACE1 is stimulated by glycosphingolipids and glycerophospholipids (83), which may regulate the location of the enzyme within membrane lipid rafts (84). Following cleavage by BACE, Aβ is generated by the action of γ-secretase. γ-secretase is an assembly of four subunits including catalytic presenilin (85) where combined units are regulated by many partners (86) including the γ-secretase activating protein (GSAP) that alters the catalytic site of presenilin to enhance APP processing (87). Notably, viral infection induces interferon-induced transmembrane protein 3 (IFITM3) to directly activate γ-secretase and elevate synthesis of Aβ (88, 89). Overall, Aβ induction is regulated at many points in response to stimuli and cell-state sensors.
In a complementary way, Aβ is reduced by multiple mechanisms. Aβ is cleared across the blood brain barrier by chaperone-mediated transport (90, 91), while cerebrospinal fluid may enter the parenchyma to flush Aβ from the CNS (92). Within the parenchyma, soluble oligomeric Aβ is engulfed into cells, and then processed by autophagy, endosomes/lysosomes and the ubiquitin-proteosome. Peripheral monocytes infiltrate the brain to produce macrophages that phagocytize Aβ, as do microglia (93). Secreted by choroid plexus, the carrier protein transthyretin may act as an Aβ scavenger to export or neutralize amyloid (94, 95). Extracellular soluble Aβ may also be catabolized by neprilysin, insulin degrading enzyme and angiotensin converting enzyme produced by neurons, microglia and astrocytes (96).
Neprilysin and transthyretin share an important feature. Their transcription is induced by the amyloid intracellular domain, which is generated when APP is cleaved by BACE (97). This action potentially provides negative feedback to regulate the quantity of extracellular Aβ. Amyloid and its precursors may also feedback upon the systems that lead to their production. The β C-terminal fragment (βCTF) generated by BACE cleavage contains a substrate inhibitory domain that negatively modulates γ-secretase (98). Within nonamyloidogenic processing of APP, the αCTF produced by α-secretase negatively modulates γ-secretase, potentially reinforcing production toward P3 and away from Aβ (99). The action of γ-secretase is also facilitated by interaction with γ-secretase activating protein (GSAP) and heat shock induction factor-1a (Hif-1a) (100, 101). Whether these proteins are inhibited in response to Aβ is unknown but if so, this could provide a further avenue for negative feedback regulation.
Overall, Aβ is balanced by mechanisms of synthesis and degradation that are mediated by negative feedback. I propose this system evolved to activate Aβ at an optimal level. Aβ entails benefits and costs, where the net effect on relative fitness (net damage) is exposed to natural selection, and this acts strongly at young ages. Thus, Aβ (and other defenses) always has its dark side but this is unavoidable in a world with pathogens. Aging, however, changes the balance.
Asymmetric regulation of Aβ in aging
Reproductive value declines with age (102) and selection acting in the old therefore has little force to maintain defense regulatory systems if they are compromised. If the mechanism of negative feedback are degraded in aged individuals, the balance between induction and feedback will not maintain Aβ at its optimum. The loss of feedback may permit incremental accumulation of Aβ when negative regulation is a continuous mechanism. Alternatively, Aβ may accumulate precipitously if the negative regulatory system is based on hysteresis and aging increases the lag-time between inductive and terminating signals. In either case, the consequence is elevated Aβ in old individuals because the negative feedback (F) does not balance the level of induction (I) (Figure 1B). We experience high defense costs of Aβ in the form of neurodegeneration even while the elevated Aβ reduces the cost of direct virulence.
Unlike the processes of amyloid clearance (103-105), little is known about how age impacts mechanisms that terminated Aβ production, that is, how a proximal mechanism of AD will be shaped by declining force of natural selection. Central to my argument, innate immune systems can be controlled through negative feedback (106-108). Two examples illustrate the potential for this concept applied to AD. Type I interferon increases in the choroid plexus of aged mice (109, 110) and contributes to elevated expression of IFITM3, where IFITM3 activates γ-secretase (88, 89). Notably, Aβ suppresses microglial myocyte enhancer factors, which are positive regulators of Type I interferon (111). Thus, Aβ has the potential to negatively regulate its own production, and where there is a lag between induction and negative repression. It would be of interest to understand if components in this feedback network are impaired with age, and whether this increases the level of Aβ. In a second example some microRNA reduce NFκB, and this subsequently down-regulates innate immunity (112, 113). Potentially, microRNA secreted in exosomes from microglia (114) may be induced by Aβ and thereby mediate production of Aβ within the brain, while aging somehow interrupts this negative regulation.
Naturally, age will also increase exposure to stimuli that initiate Aβ production. Exposure to infection is cumulative in old individuals. Age also associates with decreased blood brain barrier function that increases pathogen entry to the CNS (115, 116). As well, latent infective agents such as HSV, HCMV or Toxoplasma are reactivated with age (45, 117, 118). Mechanisms that suppress LINE-1 retrotransposition may decline with age and thereby trigger Type I interferon (53, 119). Any of these age-dependent events will elevate the magnitude of direct virulence and further induce Aβ, increasing the net damage to the host (the point Old* in Fig 1C).
The defense model of AD may explain why high levels of amyloid are found in the brains of some people without dementia (Fig 1D). They may accumulate Aβ upon infection, and even have poor negative regulation of the response, but their cost function for this Aβ defense is inherently low; they experience little net damage from Aβ (the point Old# in Fig 1D). This explanation is difficult to explore within humans, but Aβ is thought to cause little damage in the naked mole rat where levels are similar to that of 3xFAD mice (120). Likewise, nonhuman great apes accumulate Aβ but lack measurable dementia (121). Conversely, APOE-ε4 (122) may alter the shape of the Aβ defense cost function such that cost is low relative to APOE-ε3 at young ages but accelerates more rapidly at late ages. APOE-ε4 carriers at late age may experience greater neurological damage per unit of Aβ expression. This model may account for the commonness of APOE-ε4 because young carriers of APOE-ε4 would incur a lower net fitness cost of Aβ upon infection than non-carriers. Consistent with this expectation, APOE-ε4 carriers appear to be better protected against early life infection among rural Ghanaians exposed to water borne pathogens (123).
This model also organizes thinking about familial- and sporadic-AD. FAD individuals carry mutations in genes involved in Aβ production such as presenilin. Consequently, the level of induced amyloid should be elevated beyond the wildtype optimum even at young ages, as is observed in APP and PSEN carriers assessed for cerebrospinal fluid biomarkers (124). Unlike the explanation proposed for APOE-ε4, elevated Aβ in FAD carriers may be expected to have higher net cost than noncarriers. Cognitive impairment short of diagnosed AD may still impact the ability to care for children and grandchildren, and this may restrain the frequency of FAD alleles. As individuals with FAD age, their propensity to express Aβ advances the onset and penetrance of AD. Sporadic AD on the other hand arises from stochastic, age-associated events that induce defense expression (Aβ) — new, cumulative or latent infection — or impair the mechanisms of negative regulatory feedback. Unlike FAD, individuals that eventually develop SAD experience optimal levels of Aβ expression when young. The risks that induce Aβ in cases of SAD may be influenced by genetic polymorphisms, as when HSV-1 infection increases the incidence of AD among APOE-ε4 carriers (125). As well, this view suggests that polymorphisms affecting the negative regulation of innate immunity may be risk factors for SAD.
Conclusion
Aβ in good health and bad
Any negative consequences of Aβ are usually considered to arise only when the protein becomes abnormally abundant. The concept of defense optimization proposes Aβ is always costly, it is always a ‘double-edged sword’ (126). But when the activity of Aβ is at its optimum we cannot perceive its costs or benefits — we simply appear ‘normal’. We observe costs when Aβ is elevated beyond normal, when the additional costs exceed the benefits. Overall, this defense model of AD is consistent with an underlying principle of how health and disease evolve (127): ‘good health’ is a compromise where the phenotype favored by evolution is less bad than the available alternatives.
My view emphasizes how Aβ is a proximal feature of a regulated defense, where natural selection shapes this regulation. Clearly, the CNS regulates many defenses, including other antimicrobial neuropeptides (128, 129), neuroinflammation (130), and cellular senescence (131). All defense mechanisms incur costs along with their benefits, and I propose each defense will be optimized through balanced systems of induction and negative feedback. As with Aβ, the regulatory mechanisms may become asymmetric in old individuals and contribute to neurodegeneration: strong induction with weak feedback. Furthermore, if negative regulatory systems control multiple defense mechanisms, mitigating the costs of one feature alone such as by removing Aβ may not resolve AD. Excessive costs from other defense systems can still drive AD pathology. Multiple inductive stimuli may also tip the balance of Aβ in older individuals, making it difficult to identify one etiological agent (132). In a translational context I suggest a practical perspective to address AD by understanding the events that reduce negative regulation of interlocking immune defense systems.
There are precedents for this theme. Nalivaeva (96) argued Aβ levels in the brain sit at a dynamic equilibrium between production and clearance. Puzzo (133) proposed AD arises when Aβ loses its ability to exert negative feedback through induction of alpha7 nicotinic acetylcholine receptors. Wick (134) suggested inflammation initially inhibits Aβ toxicity but persistent inflammation produces pathology. Here I emphasize how selection may have optimized the costs of Aβ relative its benefits, how negative feedback regulation may stabilize this optimum and how aging releases selection upon the maintenance of this feedback.
This thesis points to practice: understand how to preserve and restore negative feedback systems that regulate Aβ activity and defense systems in general. An aged brain may then induce an appropriate defense to a pathogen even when aging increases pathogen susceptibility, and then effectively terminate the defense as it does in young individuals. This perspective recommends explicit research on how Aβ production is negatively regulated through feedback, and more broadly how innate immunity and the inflammatory response of the brain is regulated to self-terminate.
Ethical standards: The author declares ethical standards are upheld.
Funding: MT was supported by the National Institutes of Health (USAS), NIA, R21 AG067323. The sponsor had no role in the concepts or preparation of the manuscript.
Acknowledgements: MT thanks his students over the years for the opportunity to learn alongside them the concepts of Darwinian Medicine that brought this paper to life.
Conflict of interest statement: MT declares no conflict of interest.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. 2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 2022;18(4):700-897. 10.1002/alz.12638.
2. Kent SA, Spires-Jones TL, Durrant CS. The physiological roles of tau and Aß: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020;140(4):417-47. 10.1007/s00401-020-02196-w.
3. Long JM, Holtzman DM. Alzheimer disease: An update on pathobiology and treatment strategies. Cell. 2019;179(2):312-39. 10.1016/j.cell.2019.09.001.
4. Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, et al. Assembly and aggregation properties of synthetic Alzheimer’s A4/beta amyloid peptide analogs. J Biol Chem. 1992;267(1):546-54.
5. Gosztyla ML, Brothers HM, Robinson SR. Alzheimer’s amyloid-β is an antimicrobial peptide: A review of the evidence. J Alzheimers Dis. 2018;62(4):1495-506. 10.3233/jad-171133.
6. Brothers HM, Gosztyla ML, Robinson SR. The physiological roles of amyloid-β peptide hint at new ways to treat Alzheimer’s disease. Front Aging Neurosci. 2018;10:118. 10.3389/fnagi.2018.00118.
7. Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5(3):e9505. 10.1371/journal.pone.0009505.
8. Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med. 2016;8(340):340ra72. 10.1126/scitranslmed.aaf1059.
9. Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement. 2018;14(12):1602-14. 10.1016/j.jalz.2018.06.3040.
10. Nesse RM. Tinbergen’s four questions: two proximate, two evolutionary. Evol Med Public Health. 2018;2019(1):2. 10.1093/emph/eoy035.
11. Dobzhansky T. Nothing in biology makes sense except in the light of evolution. Am Biol Teacher. 1973;35(3):125-9. 10.2307/4444260.
12. Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J Neurosci. 2006;26(40):10129. 10.1523/JNEUROSCI.1202-06.2006.
13. Eimer WA, Vassar R. Neuron loss in the 5xFAD mouse model of Alzheimer’s disease correlates with intraneuronal Aβ42 accumulation and Caspase-3 activation. Mol Neurodegener. 2013;8(1):2. 10.1186/1750-1326-8-2.
14. Nakai T, Yamada K, Mizoguchi H. Alzheimer’s disease animal models: Elucidation of biomarkers and therapeutic approaches for cognitive impairment. Int J Mol Sci. 2021;22(11):5549. 10.3390/ijms22115549.
15. Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D’Avanzo C, et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature. 2014;515(7526):274-8. 10.1038/nature13800.
16. Karran E, Mercken M, Strooper BD. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10(9):698-712. 10.1038/nrd3505.
17. Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman DM, Hyman BT, et al. Alzheimer disease. Nat Rev Dis Primers. 2021;7(1):33. 10.1038/s41572-021-00269-y.
18. Karran E, De Strooper B. The amyloid hypothesis in Alzheimer disease: New insights from new therapeutics. Nat Rev Drug Discov. 2022;21(4):306-18. 10.1038/s41573-022-00391-w.
19. Panza F, Lozupone M, Logroscino G, Imbimbo BP. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2019;15(2):73-88. 10.1038/s41582-018-0116-6.
20. Decourt B, Boumelhem F, Pope ED, 3rd, Shi J, Mari Z, Sabbagh MN. Critical appraisal of amyloid lowering agents in AD. Curr Neurol Neurosci Rep. 2021;21(8):39. 10.1007/s11910-021-01125-y.
21. Perez-Nievas BG, Stein TD, Tai HC, Dols-Icardo O, Scotton TC, Barroeta-Espar I, et al. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain. 2013;136(Pt 8):2510-26. 10.1093/brain/awt171.
22. Beker N, Ganz A, Hulsman M, Klausch T, Schmand BA, Scheltens P, et al. Association of cognitive function trajectories in centenarians with postmortem neuropathology, physical health, and other risk factors for cognitive decline. JAMA Netw Open. 2021;4(1):e2031654. 10.1001/jamanetworkopen.2020.31654.
23. Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415(6870):389-95. 10.1038/415389a.
24. Nesse RM. Natural selection and the regulation of defenses. A signal detection analysis of the smoke detector principle. Evol Hum Behav. 2005;26:88-105.
25. Read AF, Allen JE. Evolution and immunology. The economics of immunity. Science. 2000;290(5494):1104-5. 10.1126/science.290.5494.1104.
26. Okin D, Medzhitov R. Evolution of inflammatory diseases. Curr Biol. 2012;22(17):R733-40. 10.1016/j.cub.2012.07.029.
27. Newton AH, Cardani A, Braciale TJ. The host immune response in respiratory virus infection: Balancing virus clearance and immunopathology. Semin Immunopathol. 2016;38(4):471-82. 10.1007/s00281-016-0558-0.
28. Zerofsky M, Harel E, Silverman N, Tatar M. Aging of the innate immune response in Drosophila melanogaster. Aging Cell. 2005;4(2):103-8. 10.1111/j.1474-9728.2005.00147.x
29. Nathan C. Points of control in inflammation. Nature. 2002;420(6917):846-52. 10.1038/nature01320.
30. Eloseily EM, Cron RQ. Macrophage activation syndrome. In: Ragab G, Atkinson TP, Stoll ML, editors. The Microbiome in Rheumatic Diseases and Infection. Cham: Springer International Publishing; 2018. p. 151-82.
31. Dunster JL, Byrne HM, King JR. The resolution of inflammation: A mathematical model of neutrophil and macrophage interactions. Bull Math Biol. 2014;76(8):1953-80. 10.1007/s11538-014-9987-x.
32. Wang G. Raison d’etre of insulin resistance: The adjustable threshold hypothesis. J R Soc Interface. 2014;11(101):20140892. 10.1098/rsif.2014.0892.
33. Serhan CN, Savill J. Resolution of inflammation: The beginning programs the end. Nat Immunol. 2005;6(12):1191-7. 10.1038/ni1276.
34. Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat Immunol. 2001;2(7):612-9. 10.1038/89759.
35. Malynn BA, Ma A. A20: A multifunctional tool for regulating immunity and preventing disease. Cell Immunol. 2019;340:103914. 10.1016/j.cellimm.2019.04.002.
36. Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science. 2000;290(5497):1768-71. 10.1126/science.290.5497.1768.
37. Dando SJ, Mackay-Sim A, Norton R, Currie BJ, St John JA, Ekberg JA, et al. Pathogens penetrating the central nervous system: Infection pathways and the cellular and molecular mechanisms of invasion. Clin Microbiol Rev. 2014;27(4):691-726. 10.1128/CMR.00118-13.
38. Labzin LI, Heneka MT, Latz E. Innate immunity and neurodegeneration. Annu Rev Med. 2018;69:437-49. 10.1146/annurev-med-050715-104343.
39. Rossi B, Constantin G, Zenaro E. The emerging role of neutrophils in neurodegeneration. Immunobiol. 2020;225(1):151865. 10.1016/j.imbio.2019.10.014.
40. Saitgareeva AR, Bulygin KV, Gareev IF, Beylerli OA, Akhmadeeva LR. The role of microglia in the development of neurodegeneration. Neurol Sci. 2020;41(12):3609-15. 10.1007/s10072-020-04468-5.
41. Itzhaki RF, Wozniak MA, Appelt DM, Balin BJ. Infiltration of the brain by pathogens causes Alzheimer’s disease. Neurobiol Aging. 2004;25(5):619-27. 10.1016/j.neurobiolaging.2003.12.021.
42. Balin BJ, Hammond CJ, Little CS, Hingley ST, Al-Atrache Z, Appelt DM, et al. Chlamydia pneumoniae: An etiologic agent for late-onset dementia. Front Aging Neurosci. 2018;10:302. 10.3389/fnagi.2018.00302.
43. Shoji H, Wakasugi K, Miura Y, Imaizumi T, Kazuyama Y. Herpesvirus infections of the central nervous system. Jpn J Infect Dis. 2002;55(1):6-13.
44. Wainberg M, Luquez T, Koelle DM, Readhead B, Johnston C, Darvas M, et al. The viral hypothesis: How herpesviruses may contribute to Alzheimer’s disease. Mol Psychiatry. 2021;26(10):5476-80. 10.1038/s41380-021-01138-6.
45. Qin Q, Li Y. Herpesviral infections and antimicrobial protection for Alzheimer’s disease: Implications for prevention and treatment. J Med Virol. 2019;91(8):1368-77. 10.1002/jmv.25481.
46. Nayeri T, Sarvi S, Sharif M, Daryani A. Toxoplasma gondii: A possible etiologic agent for Alzheimer’s disease. Heliyon. 2021;7(6):e07151. 10.1016/j.heliyon.2021.e07151.
47. Pappas G, Roussos N, Falagas ME. Toxoplasmosis snapshots: Global status of Toxoplasma gondii seroprevalence and implications for pregnancy and congenital toxoplasmosis. Int J Parasitol. 2009;39(12):1385-94. 10.1016/j.ijpara.2009.04.003.
48. Sullivan WJ, Jr, Jeffers V. Mechanisms of Toxoplasma gondii persistence and latency. FEMS Microbiol Rev. 2012;36(3):717-33. 10.1111/j.1574-6976.2011.00305.x.
49. Nayeri Chegeni T, Sarvi S, Moosazadeh M, Sharif M, Aghayan SA, Amouei A, et al. Is Toxoplasma gondii a potential risk factor for Alzheimer’s disease? A systematic review and meta-analysis. Microb Pathog. 2019;137:103751. 10.1016/j.micpath.2019.103751.
50. Lurain NS, Hanson BA, Martinson J, Leurgans SE, Landay AL, Bennett DA, et al. Virological and immunological characteristics of human cytomegalovirus infection associated with Alzheimer disease. J Infect Dis. 2013;208(4):564-72. 10.1093/infdis/jit210.
51. Itzhaki RF, Klapper P. Cytomegalovirus: An improbable cause of Alzheimer disease. J Infect Dis. 2014;209(6):972-3. 10.1093/infdis/jit665.
52. Roberson PA, Romero MA, Osburn SC, Mumford PW, Vann CG, Fox CD, et al. Skeletal muscle line-1 ORF1 mRNA is higher in older humans but decreases with endurance exercise and is negatively associated with higher physical activity. J Appl Physiol (1985). 2019;127(4):895-904. 10.1152/japplphysiol.00352.2019.
53. De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, et al. L1 drives INF in senescent cells and promotes age-associated inflammation. Nature. 2019;566(7742):73-8. 10.1038/s41586-018-0784-9.
54. Panackal AA, Williamson PR. Fungal infections of the central nervous system. Continuum (Minneap Minn). 2015;21(6 Neuroinfectious Disease):1662-78. 10.1212/con.0000000000000241.
55. Alonso R, Pisa D, Fernández-Fernández AM, Carrasco L. Infection of fungi and bacteria in brain tissue from elderly persons and patients with Alzheimer’s disease. Front Aging Neurosci. 2018;10:159. 10.3389/fnagi.2018.00159.
56. Pisa D, Alonso R, Juarranz A, Rábano A, Carrasco L. Direct visualization of fungal infection in brains from patients with Alzheimer’s disease. J Alzheimers Dis. 2015;43:613-24. 10.3233/JAD-141386.
57. Little CS, Hammond CJ, MacIntyre A, Balin BJ, Appelt DM. Chlamydia pneumoniae induces Alzheimer-like amyloid plaques in brains of BALB/c mice. Neurobiol Aging. 2004;25(4):419-29. 10.1016/s0197-4580(03)00127-1.
58. Sierra A, Paolicelli RC, Kettenmann H. Cien años de microglía: Milestones in a century of microglial research. Trends Neurosci. 2019;42(11):778-92. 10.1016/j.tins.2019.09.004.
59. Ennerfelt HE, Lukens JR. The role of innate immunity in Alzheimer’s disease. Immunol Rev. 2020;297(1):225-46. 10.1111/imr.12896.
60. Casali BT, Reed-Geaghan EG. Microglial function and regulation during development, homeostasis and Alzheimer’s disease. Cells. 2021;10(4):957.
61. Busciglio J, Gabuzda DH, Matsudaira P, Yankner BA. Generation of beta-amyloid in the secretory pathway in neuronal and nonneuronal cells. Proc Natl Acad Sci USA. 1993;90(5):2092-6. 10.1073/pnas.90.5.2092.
62. Frost GR, Li Y-M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017;7(12):170228. 10.1098/rsob.170228.
63. Bhattacharyya R, Black SE, Lotlikar MS, Fenn RH, Jorfi M, Kovacs DM, et al. Axonal generation of amyloid-β from palmitoylated APP in mitochondria-associated endoplasmic reticulum membranes. Cell Rep. 2021;35(7):109134. 10.1016/j.celrep.2021.109134.
64. Fulop T, Witkowski JM, Bourgade K, Khalil A, Zerif E, Larbi A, et al. Can an infection hypothesis explain the beta amyloid hypothesis of Alzheimer’s disease? Front Aging Neurosci. 2018;10:224. 10.3389/fnagi.2018.00224.
65. Bourgade K, Garneau H, Giroux G, Le Page AY, Bocti C, Dupuis G, et al. ß-amyloid peptides display protective activity against the human Alzheimer’s disease-associated herpes simplex virus-1. Biogerontology. 2015;16(1):85-98. 10.1007/s10522-014-9538-8.
66. White MR, Kandel R, Tripathi S, Condon D, Qi L, Taubenberger J, et al. Alzheimer’s associated β-amyloid protein inhibits influenza A virus and modulates viral interactions with phagocytes. PLoS One. 2014;9(7):e101364. 10.1371/journal.pone.0101364.
67. Pastore A, Raimondi F, Rajendran L, Temussi PA. Why does the Aβ peptide of Alzheimer share structural similarity with antimicrobial peptides? Commun Biol. 2020;3(1):135. 10.1038/s42003-020-0865-9.
68. Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, et al. Alzheimer’s disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron. 2018;99(1):56-63.e3. 10.1016/j.neuron.2018.06.030.
69. Bortolotti D, Gentili V, Rotola A, Caselli E, Rizzo R. HHV-6A infection induces amyloid-beta expression and activation of microglial cells. Alzheimers Res Ther. 2019;11(1):104. 10.1186/s13195-019-0552-6.
70. Michiels E, Rousseau F, Schymkowitz J. Mechanisms and therapeutic potential of interactions between human amyloids and viruses. Cell Mol Life Sci. 2021;78(6):2485-501. 10.1007/s00018-020-03711-8.
71. Reiss AB, Arain HA, Stecker MM, Siegart NM, Kasselman LJ. Amyloid toxicity in Alzheimer’s disease. Rev Neurosci. 2018;29(6):613-27. 10.1515/revneuro-2017-0063.
72. Ferreira ST, Klein WL. The Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol Learn Mem. 2011;96(4):529-43.10.1016/j.nlm.2011.08.003.
73. Palop JJ, Mucke L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat Rev Neurosci. 2016;17(12):777-92. 10.1038/nrn.2016.141.
74. Zaretsky DV, Zaretskaia MV. Flow cytometry method to quantify the formation of beta-amyloid membrane ion channels. Biochim Biophys Acta Biomembr. 2021;1863(2):183506. 10.1016/j.bbamem.2020.183506.
75. Arispe N, Diaz JC, Simakova O. Aβ ion channels. Prospects for treating Alzheimer’s disease with Aβ channel blockers. Biochim Biophys Acta Biomembr. 2007;1768(8):1952-65. 10.1016/j.bbamem.2007.03.014.
76. Zaretsky DV, Zaretskaia MV. Mini-review: Amyloid degradation toxicity hypothesis of Alzheimer’s disease. Neurosc Let. 2021;756:135959. 10.1016/j.neulet.2021.135959.
77. Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, et al. Extensive involvement of autophagy in Alzheimer’s disease: An immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64(2):113-22. 10.1093/jnen/64.2.113.
78. Blass JP. The mitochondrial spiral. An adequate cause of dementia in the Alzheimer’s syndrome. Ann N Y Acad Sci. 2000;924:170-83. 10.1111/j.1749-6632.2000.tb05576.x.
79. Griner SL, Seidler P, Bowler J, Murray KA, Yang TP, Sahay S, et al. Structure-based inhibitors of amyloid beta core suggest a common interface with tau. eLife. 2019;8:e46924. 10.7554/eLife.46924.
80. Zhang YW, Thompson R, Zhang H, Xu H. APP processing in Alzheimer’s disease. Mol Brain. 2011;4:3. 10.1186/1756-6606-4-3.
81. Hunt CE, Turner AJ. Cell biology, regulation and inhibition of beta-secretase (BACE-1). FEBS J. 2009;276(7):1845-59. 10.1111/j.1742-4658.2009.06929.x.
82. Wozniak MA, Itzhaki RF, Shipley SJ, Dobson CB. Herpes simplex virus infection causes cellular beta-amyloid accumulation and secretase upregulation. Neurosci Let. 2007;429(2-3):95-100. 10.1016/j.neulet.2007.09.077.
83. Kalvodova L, Kahya N, Schwille P, Ehehalt R, Verkade P, Drechsel D, et al. Lipids as modulators of proteolytic activity of BACE: Involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J Biol Chem. 2005;280(44):36815-23. 10.1074/jbc.M504484200.
84. Sakurai T, Kaneko K, Okuno M, Wada K, Kashiyama T, Shimizu H, et al. Membrane microdomain switching: A regulatory mechanism of amyloid precursor protein processing. J Cell Biol. 2008;183(2):339-52. 10.1083/jcb.200804075.
85. De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003;38(1):9-12. 10.1016/s0896-6273(03)00205-8.
86. Gertsik N, Chiu D, Li YM. Complex regulation of γ-secretase: From obligatory to modulatory subunits. Front Aging Neurosci. 2014;6:342. 10.3389/fnagi.2014.00342.
87. Wong E, Liao GP, Chang JC, Xu P, Li Y-M, Greengard P. GSAP modulates γ-secretase specificity by inducing conformational change in PS1. Proc Natl Acad Sci USA. 2019;116(13):6385. 10.1073/pnas.1820160116.
88. Bailey CC, Zhong G, Huang I-C, Farzan M. IFITM-family proteins: The cell’s first line of antiviral defense. Ann Rev Virol. 2014;1(1):261-83. 10.1146/annurev-virology-031413-085537.
89. Hur J-Y, Frost GR, Wu X, Crump C, Pan SJ, Wong E, et al. The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer’s disease. Nature. 2020;586(7831):735-40. 10.1038/s41586-020-2681-2.
90. Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, et al. Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106(12):1489-99. 10.1172/jci10498.
91. Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015;11(8):457-70. 10.1038/nrneurol.2015.119.
92. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Trans Med. 2012;4(147):147ra11. 10.1126/scitranslmed.3003748.
93. Zuroff L, Daley D, Black KL, Koronyo-Hamaoui M. Clearance of cerebral Aβ in Alzheimer’s disease: Reassessing the role of microglia and monocytes. Cell Mol Life Sci. 2017;74(12):2167-201. 10.1007/s00018-017-2463-7.
94. Ghadami SA, Chia S, Ruggeri FS, Meisl G, Bemporad F, Habchi J, et al. Transthyretin inhibits primary and secondary nucleations of amyloid-β peptide aggregation and reduces the toxicity of its oligomers. Biomacromolecules. 2020;21(3):1112-25. 10.1021/acs.biomac.9b01475.
95. Gião T, Saavedra J, Cotrina E, Quintana J, Llop J, Arsequell G, et al. Undiscovered roles for transthyretin: From a transporter protein to a new therapeutic target for Alzheimer’s disease. Int J Mol Sci. 2020;21(6):2075. 10.3390/ijms21062075.
96. Nalivaeva NN, Belyaev ND, Kerridge C, Turner AJ. Amyloid-clearing proteins and their epigenetic regulation as a therapeutic target in Alzheimer’s disease. Front Aging Neurosci. 2014;6:235. 10.3389/fnagi.2014.00235.
97. Kerridge C, Belyaev ND, Nalivaeva NN, Turner AJ. The Aβ-clearance protein transthyretin, like neprilysin, is epigenetically regulated by the amyloid precursor protein intracellular domain. J Neurochem. 2014;130(3):419-31. 10.1111/jnc.12680.
98. Tian Y, Bassit B, Chau D, Li Y-M. An APP inhibitory domain containing the Flemish mutation residue modulates γ-secretase activity for Abeta production. Nat Struct Mol Biol. 2010;17(2):151-8. 10.1038/nsmb.1743.
99. Tian Y, Crump CJ, Li Y-M. Dual role of alpha-secretase cleavage in the regulation of gamma-secretase activity for amyloid production. J Biol Chem. 2010;285(42):32549-56.10.1074/jbc.M110.128439.
100. Wong E, Liao GP, Chang JC, Xu P, Li YM, Greengard P. GSAP modulates γ-secretase specificity by inducing conformational change in PS1. Proc Natl Acad Sci U S A. 2019;116(13):6385-90. 10.1073/pnas.1820160116.
101. Villa Jennifer C, Chiu D, Brandes Alissa H, Escorcia Freddy E, Villa Carlos H, Maguire William F, et al. Nontranscriptional role of Hif-1α; in activation of γ-secretase and notch signaling in breast cancer. Cell Rep. 2014;8(4):1077-92. 10.1016/j.celrep.2014.07.028.
102. Charlesworth B. Evolution in Age-structured Populations. Second ed. Cambridge: Cambridge University Press; 1994. 300 p.
103. Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, et al. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6(2):143-50. 10.1038/72237.
104. Nalivaeva NN, Zhuravin IA, Turner AJ. Neprilysin expression and functions in development, ageing and disease. Mech Ageing Dev. 2020;192:111363. 10.1016/j.mad.2020.111363.
105. Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008;28(33):8354-60. 10.1523/jneurosci.0616-08.2008.
106. Lang T, Mansell A. The negative regulation of toll-like receptor and associated pathways. Immunol Cell Biol. 2007;85(6):425-34. 10.1038/sj.icb.7100094.
107. Azhar N, Vodovotz Y. Innate immunity in disease: Insights from mathematical modeling and analysis. Adv Exp Med Biol. 2014;844:227-43. 10.1007/978-1-4939-2095-2_11.
108. Kobayashi KS, Flavell RA. Shielding the double-edged sword: Negative regulation of the innate immune system. J Leukoc Biol. 2004;75(3):428-33. 10.1189/jlb.0703321.
109. Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science. 2014;346(6205):89-93. 10.1126/science.1252945.
110. Porritt RA, Hertzog PJ. Dynamic control of type I IFN signalling by an integrated network of negative regulators. Trends Immunol. 2015;36(3):150-60. 10.1016/j.it.2015.02.002.
111. Lu F, Wang R, Xia L, Nie T, Gao F, Yang S, et al. Regulation of IFN-Is by MEF2D promotes inflammatory homeostasis in microglia. J Inflamm Res. 2021;14:2851-63. 10.2147/jir.S307624.
112. Mann M, Mehta A, Zhao JL, Lee K, Marinov GK, Garcia-Flores Y, et al. An NF-κB-microRNA regulatory network tunes macrophage inflammatory responses. Nature Commun. 2017;8(1):851. 10.1038/s41467-017-00972-z.
113. Markopoulos GS, Roupakia E, Tokamani M, Alabasi G, Sandaltzopoulos R, Marcu KB, et al. Roles of NF-κB signaling in the regulation of miRNAs impacting on inflammation in cancer. Biomedicines. 2018;6(2):40. 10.3390/biomedicines6020040.
114. Oshiumi H. Circulating extracellular vesicles carry immune regulatory miRNAs and regulate vaccine efficacy and local inflammatory response after vaccination. Front Immunol. 2021;12:685344. 10.3389/fimmu.2021.685344.
115. Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85(2):296-302. 10.1016/j.neuron.2014.12.032.
116. Panza F, Lozupone M, Solfrizzi V, Watling M, Imbimbo BP. Time to test antibacterial therapy in Alzheimer’s disease. Brain. 2019;142(10):2905-29. 10.1093/brain/awz244.
117. Elder E, Sinclair J. Hcmv latency: What regulates the regulators? Med Microbiol Immunol. 2019;208(3-4):431-8. 10.1007/s00430-019-00581-1.
118. Sullivan WJ, Jr., Jeffers V. Mechanisms of Toxoplasma gondii persistence and latency. FEMS Microbiol Rev. 2012;36(3):717-33. 10.1111/j.1574-6976.2011.00305.x.
119. Van Meter M, Kashyap M, Rezazadeh S, Geneva AJ, Morello TD, Seluanov A, et al. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat Commun. 2014;5:5011. 10.1038/ncomms6011.
120. Edrey YH, Medina DX, Gaczynska M, Osmulski PA, Oddo S, Caccamo A, et al. Amyloid beta and the longest-lived rodent: The naked mole-rat as a model for natural protection from Alzheimer’s disease. Neurobiol Aging. 2013;34(10):2352-60. 10.1016/j.neurobiolaging.2013.03.032.
121. Finch CE, Martin GM. Dementias of the Alzheimer type: Views through the lens of evolutionary biology suggest amyloid-driven brain aging is balanced against host defense. In: Alvergne A, Jenkinson C, Faurie C, editors. Evolutionary Thinking in Medicine: From Research to Policy and Practice. Cham: Springer International Publishing; 2016. p. 277-95.
122. Parhizkar S, Holtzman DM. APOE mediated neuroinflammation and neurodegeneration in Alzheimer’s disease. Semin Immunol. 2022:101594. 10.1016/j.smim.2022.101594.
123. van Exel E, Koopman JJE, Bodegom DV, Meij JJ, Knijff P, Ziem JB, et al. Effect of APOE ε4 allele on survival and fertility in an adverse environment. PLoS One. 2017;12(7):e0179497. 10.1371/journal.pone.0179497.
124. Thordardottir S, Stahlbom AK, Ferreira D, Almkvist O, Westman E, Zetterberg H, et al. Preclinical cerebrospinal fluid and volumetric magnetic resonance imaging biomarkers in Swedish familial Alzheimer’s disease. J Alzheimers Dis. 2015;43(4):1393-402. 10.3233/JAD-140339.
125. Linard M, Letenneur L, Garrigue I, Doize A, Dartigues JF, Helmer C. Interaction between APOE4 and herpes simplex virus type 1 in Alzheimer’s disease. Alzheimers Dement. 2020;16(1):200-8. 10.1002/alz.12008.
126. Salani F, Sterbini V, Sacchinelli E, Garramone M, Bossù P. Is innate memory a double-edge sword in Alzheimer’s disease? A reappraisal of new concepts and old data. Front Immunol. 2019;10:1768. 10.3389/fimmu.2019.01768.
127. Nesse RM, Williams GC. Why We Get Sick: The New Science of Darwinian Medicine: Vintage; 2012.
128. Lee EY, Chan LC, Wang H, Lieng J, Hung M, Srinivasan Y, et al. PACAP is a pathogen-inducible resident antimicrobial neuropeptide affording rapid and contextual molecular host defense of the brain. Proc Natl Acad Sci USA. 2021;118(1):e1917623117. 10.1073/pnas.1917623117.
129. Su Y, Zhang K, Schluesener HJ. Antimicrobial peptides in the brain. Arch Immunol Ther Exp (Warsz). 2010;58(5):365-77.10.1007/s00005-010-0089-7.
130. Grubeck-Loebenstein B, Blasko I, Marx F, Trieb K. Immunization with beta-amyloid: could T-cell activation have a harmful effect? Trends Neurosci. 2000;23(3):114. 10.1016/S0166-2236(99)01541-6.
131. Baker DJ, Petersen RC. Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J Clin Invest. 2018;128(4):1208-16. 10.1172/jci95145.
132. Sochocka M, Zwolińska K, Leszek J. The infectious etiology of Alzheimer’s disease. Curr Neuropharmacol. 2017;15(7):996-1009. 10.2174/1570159×15666170313122937.
133. Puzzo D, Gulisano W, Arancio O, Palmeri A. The keystone of Alzheimer pathogenesis might be sought in Aβ physiology. Neurosci. 2015;307:26-36. 10.1016/j.neuroscience.2015.08.039.
134. Wick G, Berger P, Jansen-Dürr P, Grubeck-Loebenstein B. A Darwinian-evolutionary concept of age-related diseases. Exp Gerontol. 2003;38(1):13-25. 10.1016/S0531-5565(02)00161-4.