
M. Torso1, G.R. Ridgway1, I. Hardingham1, A.J. Schwarz2,3, S.A. Chance1 for the Alzheimer’s Disease Neuroimaging Initiative*
1. Oxford Brain Diagnostics Ltd, Oxford, UK; 2. Takeda Pharmaceuticals Ltd, Cambridge, MA, USA; 3. Current Affiliation: Qynapse Inc., Boston, MA, USA
Corresponding Author: Dr Mario Torso, Oxford Brain Diagnostics Ltd, Oxford, UK, mario.torso@oxfordbraindiagnostics.com
J Prev Alz Dis 2022;4(9):769-779
Published online June 16, 2022, http://dx.doi.org/10.14283/jpad.2022.59
Abstract
Background: Alzheimer’s disease (AD) neuropathology reveals progressive microstructural alterations of cortical architecture. Recent studies reported intriguing biphasic trajectories of cortical structural changes in the early stages of Alzheimer’s disease (AD), comprising decreased mean diffusivity (MD) and increased cortical thickness in cognitively normal amyloid-positive individuals, ahead of increases and decreases, respectively, in subsequent disease stages.
Objective: To better understand the cytoarchitectural correlates of these observations, we assessed novel cortical diffusion tensor imaging (DTI) metrics that are correlated with disruption of cortical minicolumns and protein deposition.
Design: Cross-sectional and longitudinal analysis of whole brain and temporal lobe cortical diffusivity measures. Investigation of associations between baseline cortical diffusivity values and 24-month longitudinal structural-MRI changes. Investigations of the relationships between cortical diffusivity measures and biomarkers of neuroinflammation.
Setting: Alzheimer’s Disease Neuroimaging Initiative (ADNI).
Participants: Twenty-four amyloid-negative controls
(CN-), 28 amyloid-positive controls (CN+), 46 amyloid-positive subjects with mild cognitive impairment (MCI+) and 22 amyloid-positive subjects with AD were included.
Measurements: 3DT1 and DTI scans at baseline and approximately 24-month follow-up were used to calculate cortical MD and three novel cortical diffusivity measures: the angle between the radial minicolumnar axis and the principal diffusion direction (AngleR); the diffusion components perpendicular to the minicolumns (PerpPD+), and the principal diffusion component parallel with the minicolumns (ParlPD). Cortical macrostructural measurements (cortical volume fraction and cortical thickness), were used to test the hypothesis that baseline cortical diffusivity values can predict change in structural MRI outcomes over approximately 24 months. CSF soluble TREM2 and progranulin (PGRN) concentrations were used to investigate associations with microglial activity and potentially other aspects of neuroinflammation.
Results: Cortical diffusivity metrics revealed a dependence on disease stage, with AngleR and PerpPD+ displaying biphasic relationships and ParlPD a monotonic relationship with clinical severity. The novel metrics were able to differentiate between Amyloid+ and Amyloid- controls (AngleR) and to differentiate among disease stages along the AD continuum (PerpPD+). Linear regression revealed significant associations between baseline cortical diffusivity values and subsequent 24-month longitudinal structural-MRI changes. AngleR values were significantly associated with CSF sTREM2 and PGRN concentrations.
Conclusions: Cortical diffusivity parameters reflecting minicolumnar organization and neuroinflammation may provide a sensitive and biologically interpretable measurement of cortex quality and microstructure across the AD continuum.
Key words: Alzheimer’s disease, neurodegeneration, minicolumn, intracortical diffusivity.
Abbreviations: DTI: Diffusion tensor imaging; AngleR: the angle between the radial minicolumnar direction and the principal diffusion direction; PerpPD+: the diffusion components perpendicular to the minicolumns; ParlPD: the principal diffusion component parallel with the minicolumns; GM: Grey matter; CVF: Cortical volume fraction; CT: Cortical thickness; MD: Mean diffusivity.
Introduction
The expansion of the cortical surface during development (within individuals), and across evolutionary time (between species), is largely due to proliferation and spacing of radial minicolumns of cells (1). These microscopic structures persist throughout the mature brain, where they span the 3–4 mm cortical depth with a horizontal width of approximately 50 μm. Column-like radial organization is found for cell bodies and their axonal and dendritic connections.
Alzheimer’s disease (AD) neuropathology reveals progressive microstructural alterations of cortical architecture. Two phases have been proposed for these cytoarchitectural changes: a phase of synaptic and neuropil loss whereby minicolumn width declines with age, neuroinflammatory processes and amyloid build up, followed by a phase when cell loss and columnar disintegration occurs, and dementia symptoms develop (2). Minicolumnar spacing has been shown to relate to cognitive ability and was proposed as a sensitive biomarker of AD (3). Medial temporal lobe minicolumn thinning relates to MMSE (Mini-Mental State Examination) and dorsolateral prefrontal cortex minicolumns relate to IQ decline in AD (4). Similar structure-function relationships have been shown in cognitively impaired monkeys (5). In dementia with Lewy bodies, minicolumn changes were more sensitive than other common histological measures like cell density (6).
Diffusion-weighted magnetic resonance imaging (dMRI) enables in vivo assessment of brain microstructure. dMRI has been applied extensively to white matter; in cortical grey matter (GM), mean diffusivity (MD) and free water (FW) have revealed changes in water mobility accompanying changes in macrostructure (e.g. volume or thickness) in dementia (7-9). In AD specifically, clinical stages (mild cognitive impairment (MCI) or mild dementia) are characterized by both decreased GM volume/thickness and increased MD/FW. In earlier stages of disease, where amyloid plaques have accumulated but cognition is not yet impaired, intriguing increases in GM volumes and accompanying decreases in MD and FW have been observed (8), suggesting biphasic trajectories of both cortical macrostructure and microstructure over the disease course.
Diffusion models that are more directly informed by the brain’s cellular structure on the length scale probed by dMRI (~microns) may provide parameters that are more sensitive to these disease-associated changes. Some models, [NODDI (10-11), or DBSI (12)], require multi-shell dMRI protocols and/or strong diffusion gradients that are not widely available or standardized. An alternative approach, cortical disarray measurement, models cortical minicolumnar organization using single-shell dMRI. Previous studies showed that parameters of this model relate to amyloid and tau pathology in AD (13), can distinguish AD from posterior cortical atrophy (14) and subtypes of frontotemporal degeneration (15-16), and are related to histopathological measures of minicolumn alterations at autopsy (17).
The primary aim here was to assess cortical minicolumnar alterations using this model across the AD spectrum from cognitively normal to mild dementia, and associations with macrostructural changes. Given the aforementioned observations of decreased MD and increased volumes in GM, we looked at changes in the earliest pathological stages associated with amyloid accumulation prior to cognitive disfunction. A secondary aim was to evaluate longitudinal changes in cortical diffusivity measures over 24 months. The third aim was to investigate whether cortical diffusivity metrics related to neuroinflammation; more specifically, to CSF soluble TREM2 (a marker of microglial activation), and to CSF PGRN (which relates to microglial activity and other neuroinflammatory processes).
Methods
Participants
Data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public-private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimer’s disease (AD). For more information, please see www.adni-info.org.
All the cases available at the time of data extraction (January 2021) from the ADNI dataset with 3D T1 and DTI scans, amyloid PET SUVR values, clinical and demographic data at baseline and approximately 24 months later were considered for the present study. To focus on the AD continuum, amyloid negative MCI and AD participants were excluded; both amyloid negative and amyloid positive controls were included.
Amyloid positivity was determined based on florbetapir PET scans, analyzed by the ADNI PET core (UC Berkeley) with positivity determined as whole brain standardized uptake value ratio (SUVR) > 1.11. The clinical inclusion criteria for the groups included: CN subjects showed no signs of depression, mild cognitive impairment or dementia; MCI subjects had reported memory complaints, abnormal memory performance at neuropsychological evaluation, mini-mental state examination (MMSE) score between 24 and 30 (inclusive), a Clinical Dementia Rating (CDR) of 0.5 and general cognition and functional performance sufficiently preserved such that a diagnosis of AD cannot be made at the time of the screening visit. Participants with AD were those who met the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS/ADRDA) criteria for probable AD (18).
Data from a total of 120 subjects were analyzed. These comprised 52 cognitively normal controls (CN), of whom 24 were amyloid-negative (denoted CN-) and 28 were amyloid positive (CN+), 46 amyloid-positive subjects with mild cognitive impairment (MCI+) and 22 amyloid-positive AD (AD+).
Eight scans (3 CN+, 3 MCI+ and 2 AD+) that failed during processing were not included in longitudinal analyses.
Structural MRI analyses
The 3D T1-weighted images for each subject were segmented using FreeSurfer v 6.0 (https://surfer.nmr.mgh.harvard.edu/). This provided outputs containing estimates of the cortical grey matter volume (CV), hippocampal (HV) volume, and the cortical thickness (CT).
The two hippocampal volumes obtained (left and right) were averaged. To account for subjects’ head size differences, all volumes were expressed as a percentage of the total intracranial volume (ICV), namely Cortical volume fraction (CVF) and bilateral Hippocampal Volume Fraction (HVF).
Cortical diffusivity analysis
All Diffusion-weighted images were preprocessed using FSL tools (FSL Version 6.0; FMRIB Software Library, Oxford, UK—https://www.fmrib.ox.ac.uk/fsl/). Diffusion-weighted images were corrected for motion and eddy current effects by alignment of all images to a reference b = 0 image using FSL’s eddy tool. The diffusion tensor was then calculated with the FSL DTIFIT tool, providing fractional anisotropy (FA), mean diffusivity (MD) and V1 maps. For each subject, the displacement among diffusion volumes was estimated using the eddy output to obtain a measure of head motion (defined as “head motion”) during the acquisition.
Standard diffusivity analysis was conducted to calculate MD in the cortex (restricted to relatively pure cortical tissue with high confidence of being GM). Each voxel’s contribution to the mean was weighted by its calculated percentage of GM. Eg, a voxel with a predicted GM percentage of 50% counted half as much to the mean MD as a voxel predicted to be 100% GM.
Further cortical diffusivity analysis was performed using a proprietary software tool (patent application WO2016162682A1). The tool generates cortical profiles, providing an estimate of the columnar axis within the cortex. Values for the diffusion tensor derived metrics were averaged along the cortical profiles, throughout cortical GM (13, 17). Briefly, three measures were calculated, relating to the components of diffusion: AngleR was the angle between the radial minicolumn axis and the principal diffusion direction (in radians); PerpPD+ combined the components perpendicular to the radial minicolumns (× 10-3 mm2/s), and ParlPD was the principal diffusion component parallel with the radial minicolumns (× 10-3 mm2/s). PerpPD+ used here was a variant of PerpPD published previously in Torso et al (13) which includes multiple components orthogonal to the cortical columnar profile.
The temporal lobe value for each metric was the average from the combination of the following Dessikan-Killiany cortical regions: bankssts (Banks of the Superior Temporal Sulcus), entorhinal, fusiform, inferiortemporal, middletemporal, parahippocampal, superiortemporal, temporalpole, and transversetemporal.
Harmonisation
The scans included in the present study were from 16 different ADNI sites. To reduce the impact of different acquisition protocols, the data were harmonized using ComBat, an open-source library for batch-effect correction which has been used with DTI data in several published papers (19). ComBat (from “combining batches”), a location and scale model (mean & variance) uses an Empirical Bayes framework to equalize parameters of each batch while retaining group differences.
Neuroinflammation measurements
For a subcohort of 68 participants (14 CN-, 12 CN+, 24 MCI+ and 18 AD+), CSF soluble triggering receptor expressed on myeloid cells-2 (sTREM2) and progranulin (PGRN) concentrations were available in the ADNI dataset.
CSF sTREM2 and PGRN were measured by ELISA protocols previously established by the Haass group using the MSD Platform comprehensively described in previous publications (20-23). CSF levels corrected by plate-specific correction factors were used in the present study.
Statistical analyses
Data were analyzed using IBM SPSS Statistics version 26 (SPSS, Chicago, IL). Normality was tested using Shapiro-Wilk tests. The one-way Analysis of variance (ANOVA) was used to compare clinical and demographic variables. Sex and APOE ε4 carrier differences were tested with χ2-tests. These p-values were adjusted with false discovery rate correction (FDR <0.05) (24).
General Linear Model (GLM) repeated measures analyses were used to investigate cross-sectional and longitudinal differences. Cortical diffusivity (AngleR, PerpPD+, ParlPD and MD) and structural (CVF and CT) MRI measures from both time-points were entered in the model as dependent variables, with factors for the time-point (baseline or follow-up) and the diagnostic group (CN-, CN+, MCI+ or AD+), and with the baseline age as a covariate. The interaction between group and age was used to investigate the potential between-subjects differences, and the main effect of time-point (baseline – follow-up) to investigate within-subjects differences.
Differences between groups were assessed using the estimated marginal means of the fitted models, adjusted for multiple comparisons using Bonferroni’s correction.
Linear regression was used to investigate the relationship between whole brain cortical microstructural (AngleR, PerpPD+, ParlPD) and macrostructural (CVF and CT) measurements, testing the hypothesis that baseline cortical diffusivity values can predict change in structural MRI outcomes over approximately 24 months. The change in each structural MRI metric was computed as Δ = (follow up value – baseline value) / (baseline-value x time-interval). The deltas of the macrostructural measure were used in two different regression models as dependent variables and the cortical microstructural measurements (AngleR, PerpPD+, ParlPD) were entered together as predictor factors. To investigate if the potential association between micro and macrostructural measures is driven by other variables, we adjusted for age, sex and disease stage. The F-test, partial eta-squared and p-value were computed to investigate the significance of the cortical microstructural predictors adjusting for the other variables (equivalent to comparing models with and without the microstructural predictors, in each case with the other variables)
General Linear Model univariate analyses were used to investigate cross-sectional differences in baseline CSF sTREM2 and PGRN concentrations with baseline age as a covariate. The interaction between group and age was used to investigate the potential between-subjects differences. Differences between groups were assessed using the estimated marginal means of the fitted models, adjusted for multiple comparisons using Bonferroni’s correction.
Pearson partial correlation analysis, with the baseline age as a covariate, was used to investigate the association between AngleR (which had a distinctive pattern of between-group differences consistent with other findings that were considered potentially sensitive to neuroinflammation) and CSF (sTREM2 and PGRN) concentrations. Bonferroni’s correction was used to adjust for multiple comparisons.
Results
Participants
Demographics and clinical characteristics of subject groups are summarized in Table 1. There were no significant differences in age or years of education. Sex ratio was not completely balanced across groups. There were significantly more men than women in the MCI+ group compared to the CN+ group. APOE ε4 carriers were significantly less frequent in both CN groups compared to MCI+ and AD+.
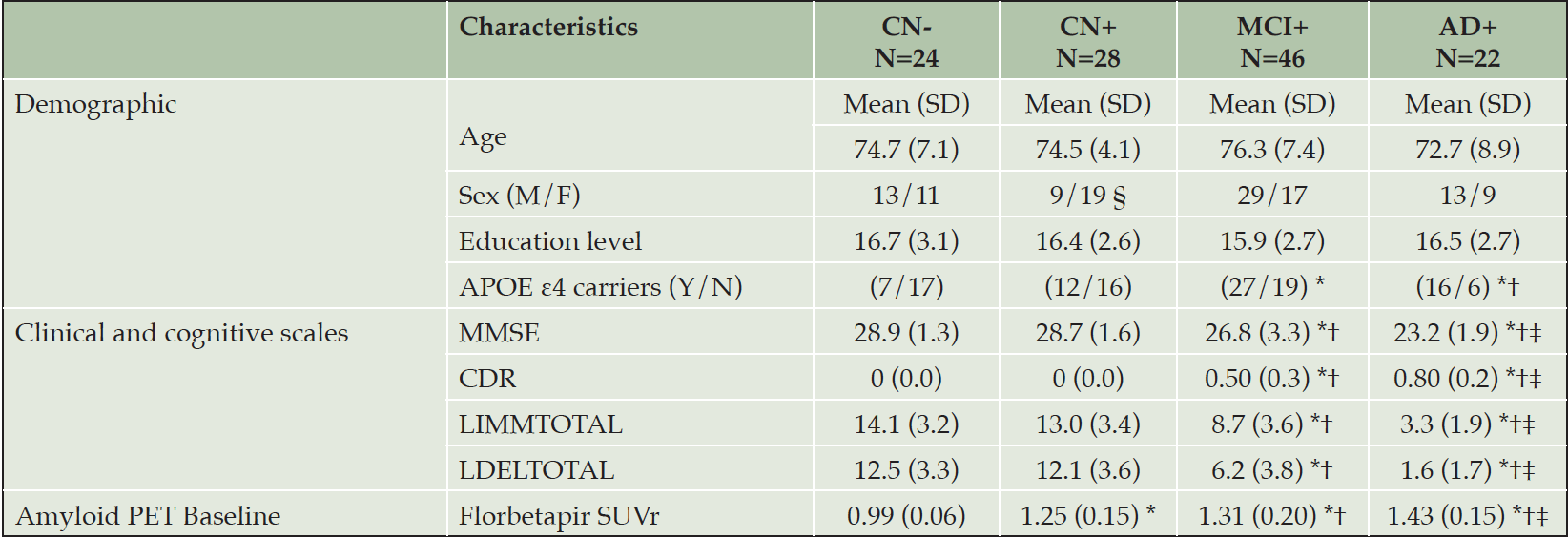
Table 1. Participant characteristics at baseline
CN- cognitively normal amyloid-beta negative, CN+ cognitively normal amyloid-beta positive, MCI+ mild cognitive impairment amyloid-beta positive, AD+ Alzheimer’s disease amyloid-beta positive, SD standard deviation, CDR clinical dementia rating scale – global score, MMSE mini-mental state examination, LIMMTOTAL Logical Memory – Immediate Recall, LDELTOTAL Logical Memory – Delayed Recall. SUVr standardized uptake value ratio; All significant differences survived after false discovery rate correction (FDR <0.05). * Significantly different compared to CN-; † Significantly different compared to CN+; ‡ significantly different compared to MCI.
The control groups performed significantly better on the MMSE and logical memory (immediate and delayed recall) than MCI+ patients, who in turn scored significantly better than the AD+ group. Mean CDR scores were significantly higher in the AD+ group, followed by the MCI+ group (Table 1). As expected, the florbetapir PET SUVr values confirmed significant differences between CN- and all the other groups. In addition, the group comparisons revealed significant differences between CN+ group and both patients’ groups (MCI and AD) and between MCI and AD groups.
Macrostructural measurements
Structural MRI measures for each group are summarized in Table 2. For all measurements, there was a monotonic decrease in mean values across diagnostic groups (CN- > CN+ > MCI+ > AD+), and the mean values in all groups were lower at 24-month follow-up than baseline. Overall, macroscopic brain atrophy measurements showed a non-linear dependence on disease stage, with differences between AD+ and MCI+ greater than those between other adjacent groups.
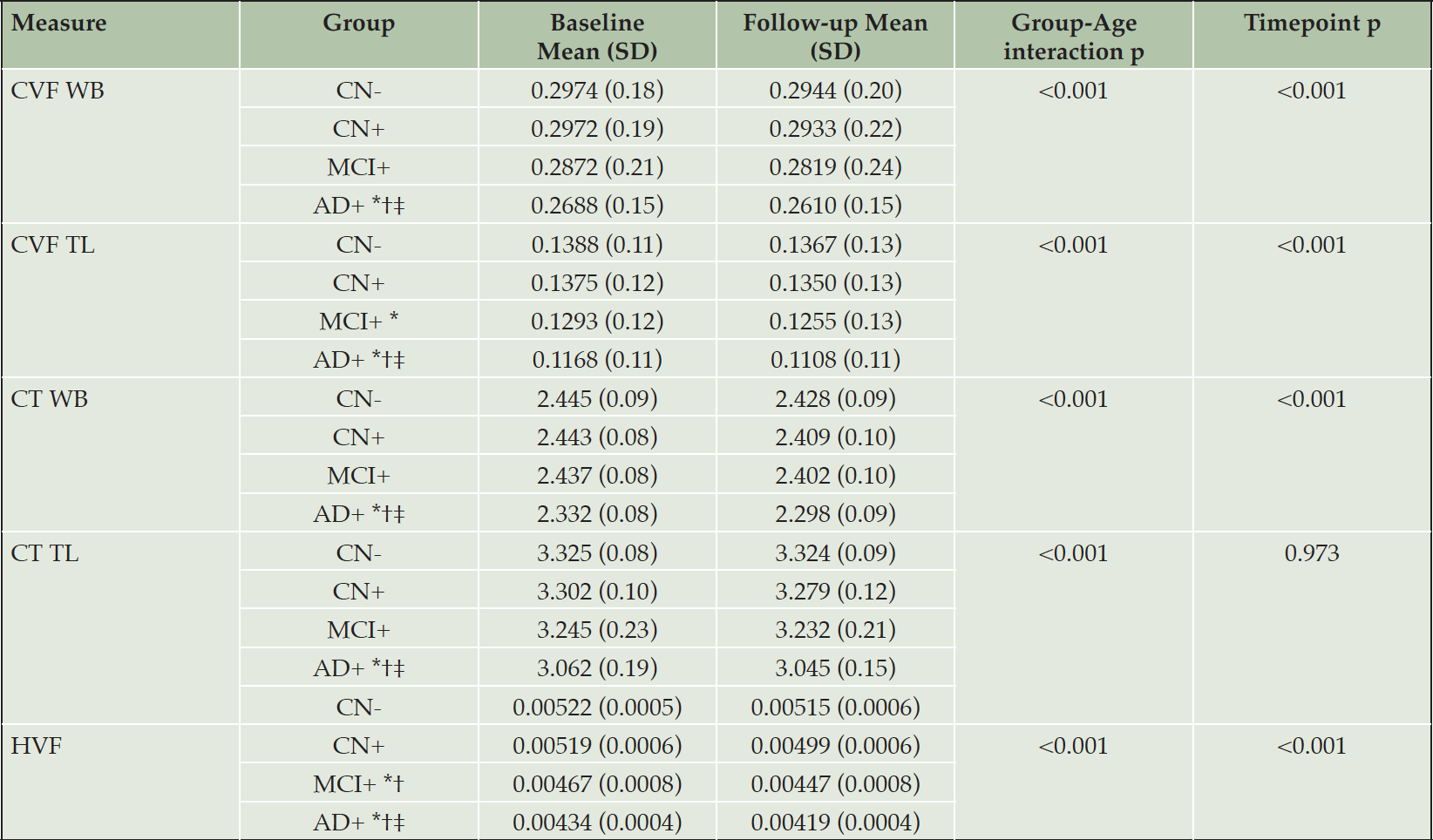
Table 2. MRI structural characteristics
CN- cognitively normal amyloid-beta negative, CN+ cognitively normal amyloid-beta positive, MCI+ mild cognitive impairment amyloid-beta positive, AD+ Alzheimer’s disease amyloid-beta positive, SD standard deviation, CT cortical thickness, CVF cortical volume fraction, HVF Hippocampal fraction, WB whole brain, TL temporal lobe.* Significantly different compared to CN-; † Significantly different compared to CN+; ‡ significantly different compared to MCI.
The GLM repeated measures analyses revealed a significant effect of the interaction diagnostic group*age in all the structural MRI measures (all p-values<0.01).
At a whole brain level, the pairwise comparisons showed a significantly lower CVF in AD than CN- (p < .0001), CN+ (p < .0001) and MCI (p < .0001) as well as a lower CT in AD compared to CN- (p < .0001), CN+ (p < .0001) and MCI (p < .001).
At a temporal lobe level, the pairwise comparisons showed a significantly lower CVF in AD than CN- (p < .0001), CN+ (p < .0001) and MCI (p < .0001). The CVF in MCI group was significantly lower compared to CN- (p < .010). The CT was significantly lower in AD than CN- (p < .0001), CN+ (p < .0001) and MCI (p < .001).
As expected, the AD group showed a significantly lower HVF than CN- (p < .0001) and CN+ (p < .0001). The HVF in MCI was significantly lower than CN- (p < .005) and CN+ (p < .05). No significant differences between MCI and AD group in HVF values were detected.
Significant differences between timepoints were detected for all the structural MRI measures considered (all p<0.001) except temporal lobe CT.
Whole brain cortical diffusivity
At a whole brain level, in contrast to the macrostructural measurements, the cortical disarray metrics revealed a more complex dependence on disease stage, with AngleR and PerpPD+ displaying biphasic relationships and ParlPD a monotonic and roughly linear relationship with clinical severity (Fig 1). Both AngleR and PerpPD+ were decreased in value in the CN+ group relative to CN-, but showed the same level (AngleR) or increased (PerpPD+) in MCI+ relative to CN-, before showing further increased mean values in the AD+ group. Values of MD, relative to CN-, were decreased in CN+ and AD+, but similar in MCI+.
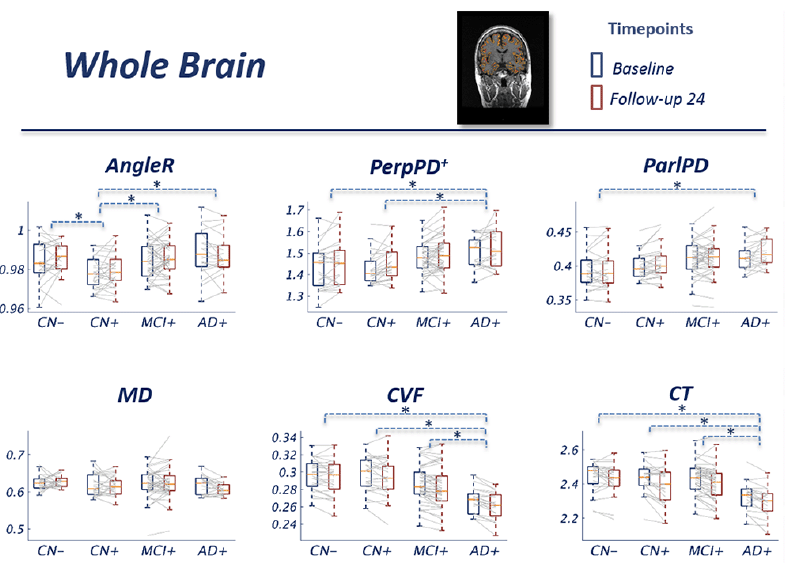
Figure 1. Whole brain cortical values
Figure 1 Group comparisons show the pattern across the diagnostic continuum using whole brain cortical diffusion measures. The blue boxes indicate the group median and interquartile range (IQR) at baseline; the red boxes indicate the group median and IQR at 24 month follow-up. Image inset shows an example MRI image illustrating cortical diffusivity measures over the segmented cortical ribbon.
The GLM repeated measures analyses revealed a significant effect of the interaction diagnostic group*age in all the diffusion measures (all p-values<0.01) except MD.
The pairwise comparisons for whole-brain values showed a significantly lower AngleR in CN+ than CN- (p<0.05), MCI+ (p<0.05) and AD+ (p<0.01) (Fig 1). PerpPD+ values were significantly higher in AD+ than CN- (p<0.05) and CN+ groups (p<0.01). ParlPD values were significantly higher in AD+ than CN- group (p<0.01). Significant differences between timepoints were detected for PerpPD+ and ParlPD (p<0.005).
Temporal Lobe cortical diffusivity
Results for metrics restricted to the temporal lobes were similar overall to those observed at the whole brain level, with AngleR exhibiting the clearest biphasic dependence on disease stage (Fig 2). The profile of PerpPD+ was attenuated relative to the whole brain results, with mean value stable in CN+ relative to CN- before increasing in later disease stages.
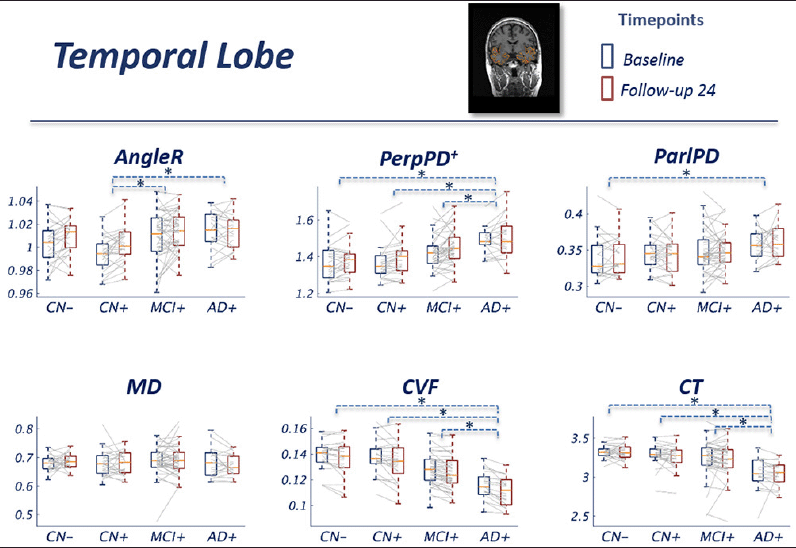
Figure 2. Temporal lobe cortical values
Figure 2 Group comparisons show the pattern across the diagnostic continuum using temporal lobe cortical diffusion measures. The blue boxes indicate the group median and IQR at baseline; the red boxes indicate the group median and IQR at 24 month follow-up. Image inset shows an example MRI image illustrating cortical diffusivity measures over the segmented temporal lobe.
The GLM repeated measures analyses revealed a significant effect of the interaction diagnostic group*age in all the diffusion measures (all p-values<0.01) except MD. Pairwise comparisons showed a significantly lower AngleR in CN+ than MCI+ (p<0.005) and AD+ (p<0.005) (Fig 2). PerpPD+ values were significantly higher in AD group than CN- (p<0.0001), CN+ (p<0.0001) and MCI+ (p<0.05). ParlPD values were significantly higher in AD than CN- (p<0.05). Significant differences between timepoints were detected for AngleR, PerpPD+ and ParlPD (p<0.005).
Micro/Macrostructural associations
Linear regressions were calculated to predict MRI structural (CVF and CT) changes (longitudinal percentage changes from baseline) using baseline cortical diffusivity whole brain measures.
All the whole brain cortical diffusivity measures, except MD, explained variance of CVF and CT changes.
Grouped together, AngleR, PerpPD+ and ParlPD explained 12.3% (F=5.039; p=0.003) of the variance in CT changes, and 14.8% (F=6.271; p=0.001) of the variance in CVF changes.
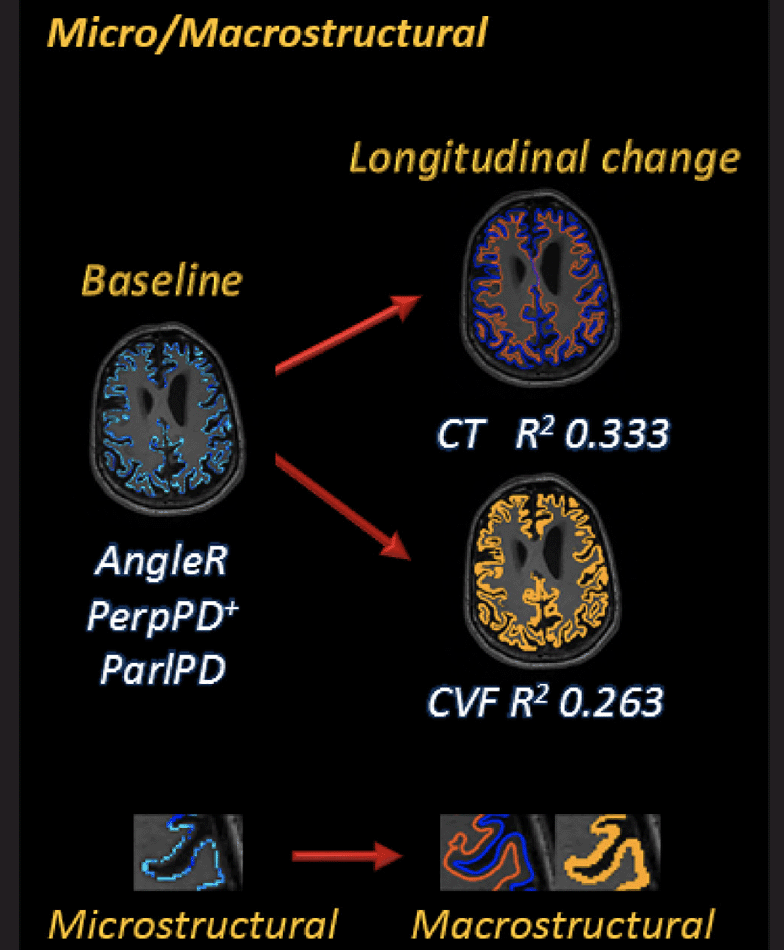
Figure 3. Micro/Macrostructural relationship
Figure 3 shows the association between baseline cortical diffusivity measures and Deltas [Δ = (24 months follow up value – baseline value) / (baseline-value x time-interval)] of structural MRI measures investigated using linear regression models. The magnifications show the characteristics of each GM cortical measure.
Comparing regression models with and without cortical microstructural values as predictors, the results confirmed the significance of the cortical microstructural predictors for CT (F=3.256; ηp2=0.086; p=0.024) and CVF (F=4.189; ηp2=0.108; p=0.007) changes.
Microglial activation and neuroinflammation
The GLM univariate analyses revealed a significant effect of the interaction diagnostic group*age in CSF PGRN concentration (p-values<0.05). Qualitatively, the dependence of PGRN concentration on disease stage (Figure 4) mirrored that of AngleR observed in the whole brain and in the temporal lobe (Figures 1 & 2). Post hoc pairwise comparisons showed PGRN levels were significantly different between CN+ and MCI+ (p<0.05) and between CN+ and AD+ (p<0.01) (Fig 4). No significant effects were detected in sTREM2 concentration (Fig 4). The levels of the two CSF neuroinflammatory measures were significantly correlated, when all diagnostic categories were combined (r=0.409; p=0.001).
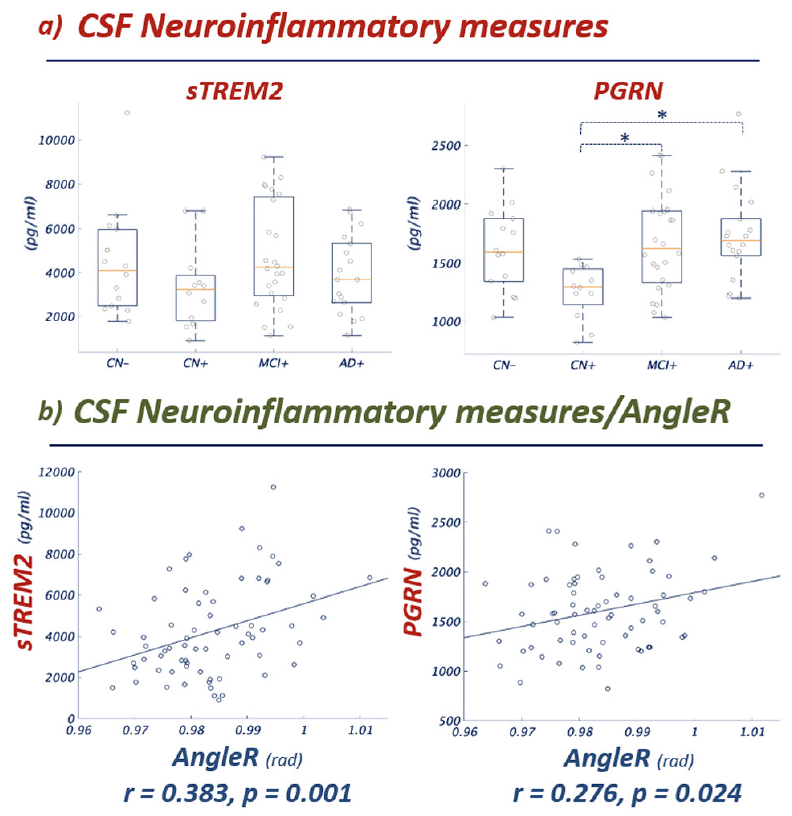
Figure 4. Micro/Macrostructural relationship
Figure 4 Panel a) Group comparisons show significant differences between groups in CSF sTREM2 and PGRN concentration (the pattern between groups is notably similar to the pattern shown for AngleR in Figures 1 and 2). Panel b) Partial Pearson correlations between CSF sTREM2 and PGRN values and AngleR. Both correlations remained significant after Bonferroni correction (0.05/2). sTREM2 and PGRN are expressed in pg/ml. AngleR is expressed in radians.
To further investigate the association between CSF neuroinflammatory measures and the AngleR cortical diffusivity measure, a partial correlation analysis, with the baseline age as a covariate, was performed. The results revealed significant positive associations between whole brain AngleR values and sTREM2 levels (r= 0.383; p=0.001) and between whole brain AngleR values and PGRN levels (r= 0.276; p=0.024). Both associations remained significant after Bonferroni correction (0.050/2). To investigate the potential influence of beta amyloid and tau CSF concentrations, two additional correlation analyses were performed, adjusting for age and CSF Aβ42 in the first analysis, then age and CSF pTau in the second analysis. Both analyses confirmed the significance of the associations between AngleR values and sTREM2 levels and between AngleR values and PGRN levels after adjusting for beta amyloid and tau. The results remained significant after Bonferroni correction.
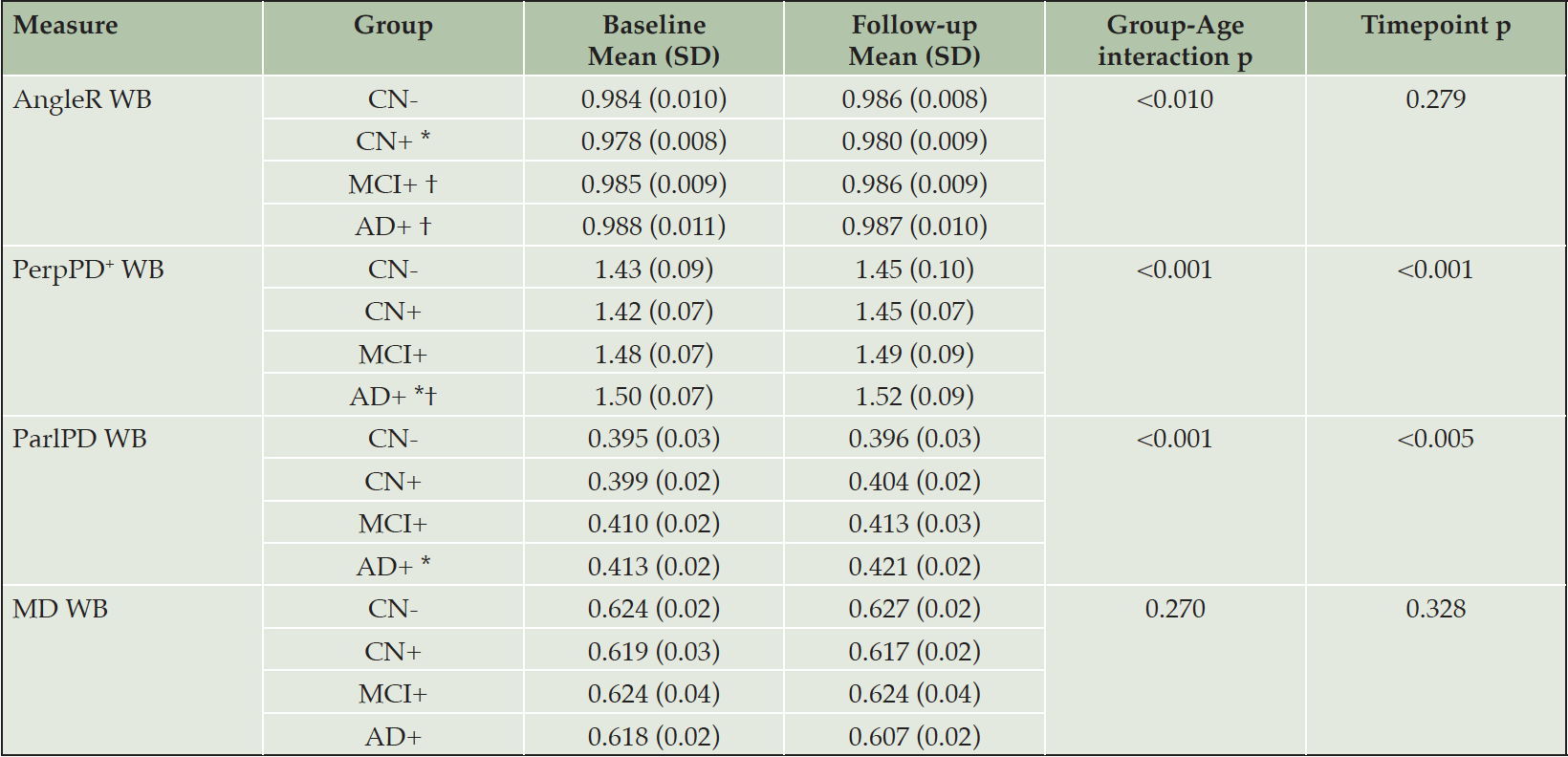
Table 3. Whole brain cortical diffusivity values
CN- cognitively normal amyloid-beta negative, CN+ cognitively normal amyloid-beta positive, MCI+ mild cognitive impairment amyloid-beta positive, AD+ Alzheimer’s disease amyloid-beta positive, SD standard deviation, WB whole brain. * Significantly different compared to CN-; † Significantly different compared to CN+; ‡ significantly different compared to MCI.
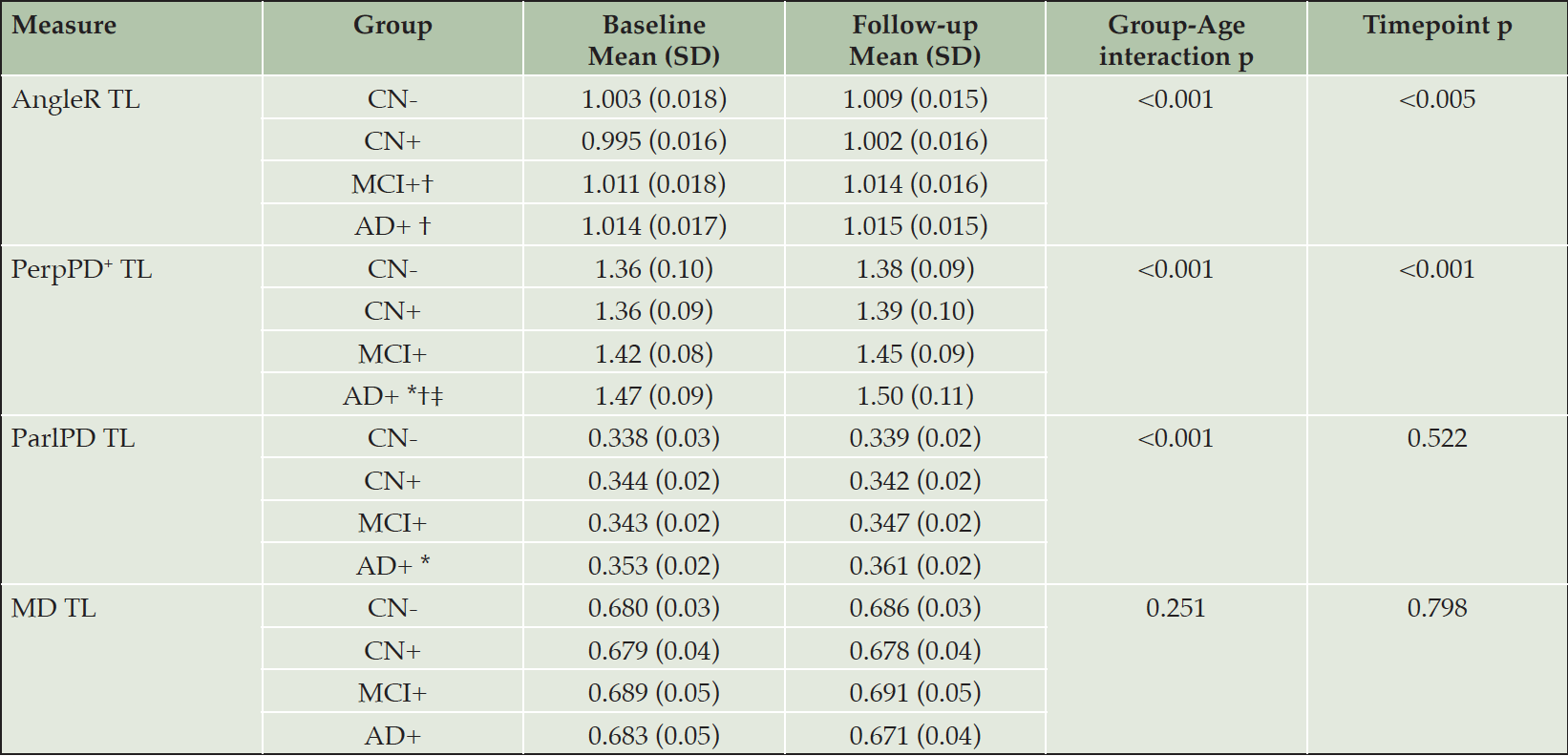
Table 4. Temporal lobe cortical diffusivity values
CN- cognitively normal amyloid-beta negative, CN+ cognitively normal amyloid-beta positive, MCI+ mild cognitive impairment amyloid-beta positive, AD+ Alzheimer’s disease amyloid-beta positive, SD standard deviation, TL temporal lobe. * Significantly different compared to CN-; † Significantly different compared to CN+; ‡ significantly different compared to MCI.
Discussion
In the present study, the cortical columnar organization signal across the AD continuum was investigated in vivo using a set of novel cortical diffusivity measures. In particular, the AngleR parameter, reflecting the angle between the principal diffusion direction and the minicolumnar direction, was found to be the microstructural metric most sensitive to the biphasic profiles previously reported, in which transient changes in the CN+ group run directionally counter to the effects of neurodegeneration in more severe disease. These findings expand on previous studies of cortical diffusivity, in which biphasic trajectories of MD (computed using surface-based analysis pipelines) and cortical thickness have been observed in temporal and parietal regions (8, 9), and these novel minicolumnar diffusivity metrics have been shown to be elevated in AD relative to healthy controls, and to correlate with biomarkers of amyloid and tau (13).
Neuropathology studies have shown that AD pathology produces profound minicolumn changes (2, 3). More specifically, a two-phase model has been proposed for these cytoarchitectural changes: a phase of synaptic and neuropil loss whereby minicolumn width declines with age as part of normal ageing, involving increasing demand for compensatory plasticity, amyloid build up and reactive microgliosis and astrogliosis, and then a later phase that lies past a threshold beyond which cell loss and columnar disintegration occur and the symptoms of dementia develop (2). Across these phases, the balance of anatomical components which dominantly influence the DTI signal may shift, resulting in different trajectories of progression for different micro- and macrostructural outcome measures. That is, the direction or linearity of the relationships of these variables and clinical severity (and between the different structural variables themselves), may vary across the AD continuum.
In this study, while PerpPD+ shows a consistent increase with increasing severity, for AngleR in particular, there is an indication of a non-monotonic progression, reminiscent of recent reports of a biphasic dependence of cortical mean diffusivity on disease severity in both familial and sporadic AD (8, 9). Those previous reports suggested inflammatory processes may be contributing to the biphasic pattern. A similar effect was not seen here for the standard MD measure. One possible reason is that the Montal findings (on a different cohort) were localised to relatively small regions or sub-regions in the temporal and parietal lobes, identified by vertex-wise analyses, whereas in the present study, the whole brain and whole temporal GM regions were pre-specified, such that weaker or more localized MD effects may have been diluted. Another potential contributor is that our standard MD measure is a (deliberately) basic voxel-based measure, while both the MD measure in Montal et al. and our novel measures use more sophisticated vertex-based or cortical-profile-based approaches, respectively. Moreover, in a previous study (13) MD showed relatively high classification accuracy in a cohort acquired on a single scanner but showed lower accuracy in a multicentre cohort (ADNI). However, AngleR did detect a biphasic-like progression in the present study and taken together with these previous reports, our findings provide further evidence in support of biphasic trajectories of structural neurodegeneration markers in early stages of AD, to which microstructural metrics seem particularly sensitive. Moreover, our findings suggest that these novel cortical diffusivity metrics may be more sensitive and better able to detect changes at the whole cortex and lobar level, amenable to pre-specification in clinical trials.
The chronology for different inflammatory processes is challenging to disentangle as some may stimulate both Amyloid and Tau accumulation, for example, the NLRP3 inflammasome has been shown to be essential for progression of amyloid Beta (Aβ) pathology in mice and also triggers hyperphosphorylation of Tau (25). While inflammatory responses are associated with, and contribute to, the development of tau deposits (accelerating the disease course) misfolded tau may, in turn, also be a trigger for microglial activation (26). However, this is likely be later in the disease process, whereas the inflammatory response linked to amyloid accumulation is expected to be earlier in the disease – in mice, synaptic deficit occurs within 3 weeks of amyloid deposition (27) creating an early inflammatory trigger. Amyloid deposition is considered to be an inducer of the chronic inflammatory response driven by activated microglia and astrocytes (28). The consequence of this inflammopathic loop is neurodegeneration – synaptic dysfunction is common to both amyloid and tau inflammatory pathways. As with synaptic changes due to amyloid deposition, synapse loss occurs early in tauopathies and is the strongest correlate of cognitive decline even when amyloid is not a significant contributor (29).
In the present study, the relationship between AngleR and neuroinflammatory processes reactive to β-amyloidosis was investigated using CSF sTREM2 and PGNR concentrations. In sporadic AD, the β-amyloidosis is mainly due to an imbalance between production and lack of clearance of Aβ peptides (30). Reactive astrocytes and activated microglial cells are commonly associated with dense-core amyloid plaques, indicating that Aβ is a major trigger of this glial response (31).
Previous studies showed that TREM2 and PGRN are well-established regulators of microglia during neurodegeneration (32, 33). The TREM2 gene is responsible for sustaining microglia phagocytosis of Aβ deposits (34, 35) and its up-regulation around amyloid plaques reflects a defence mechanism to remove protein deposition (36) by creating a barrier around the plaque (37). Its expression increases during plaque spreading and CSF sTREM2 levels go up as plaque load increases (30). However, as shown in a mouse model of AD, in some TREM2 mutations, alterations of microglial amyloid receptors cause a deficit in detection and phagocytic response to Aβ plaques during plaque development (36, 38). Reduced phagocytosis may prevent the engulfment of cellular debris, which could result in a chronic inflammatory response (20). In addition, the haplodeficiency of the microglial gene TREM2 can impair the protective function of the microglia barrier leading to decreased amyloid plaque compaction and severe axonal dystrophy (39). TREM2 alterations impairing cell surface transport and phagocytosis can lead to reduced CSF concentration of soluble TREM2 in AD patients (20).
Recent evidence suggests that CSF sTREM2 levels change dynamically with pathological processes (40) with a decrease in CSF sTREM2 in the early pre-clinical stage of Aβ pathology and a progressive increase with the tau deposition and neurodegeneration. Other observations have shown that CSF sTREM2 is associated with clinical progression rate in AD patients, with higher CSF sTREM2 concentration associated with slow clinical progression, indicating a protective role of microglia (41). Neuropathologically, this potential neuroprotective role of microglia in early stages of plaque development, may be due to the fact that areas covered by microglial processes develop fewer dystrophic neurites (39).
As shown by previous studies, PGRN is strongly increased in microglia around amyloid plaques (42) and its elevated expression reduces the amyloid plaque burden (43). CSF PGRN increases in the course of AD progression and is associated with sTREM2 (44). Consistent with previous findings (40, 44), in the studied cohort we found a reduced CSF PGRN and sTREM2 level in the cognitively unimpaired amyloid positive (CN+) group, and a progressive increase across the AD continuum towards greater severity. The two CSF measures were significantly associated (44). One possible interpretation of this dynamic progression is that microglia initially form a barrier around the plaque, sTREM2 and PGRN released by microglia remain in the plaque until the barrier fails, followed by increased concentration in the CSF of sTREM2 and PGRN (37, 45). In the later stages, the TREM2 and PGRN concentrations further increase and are associated with tau deposition and neural injury (41, 46-47). The amyloid deposition, microglial activation, reduced phagocytosis, TREM2, and PGRN deposition around the plaques may also be linked to an altered diffusivity in the cytoarchitectural spacing between minicolumns, detected by AngleR.
An association between cortical diffusivity measures and astrocytosis in autosomal-dominant Alzheimer disease was shown previously (48). The observed decreases in whole brain and temporal lobe AngleR values in amyloid-positive compared with amyloid-negative controls could thus reflect underlying neuroinflammatory mechanisms in early stages of the disease, while the significantly increased values in MCI and AD groups could reflect neuronal loss and columnar disintegration in later phases of the disease.
The significant difference between cognitively normal groups in AngleR in the absence of structural differences (CVF, HVF or CT), supports the idea that early microstructural changes related to amyloid deposition may occur in normal-appearing brain tissue. In addition, the precedence of microstructural changes is also supported by the ability to predict later atrophy from baseline microstructure. Microstructure may thus provide a sensitive early marker of AD. In contrast, the monotonic increase of PerpPD+ along the AD continuum matches the expected spread of the underlying pathological alterations and the cognitive performance, suggesting that PerpPD+ could be a useful measure of disease severity.
The findings here reinforce and expand on recent reports of microstructural cortical changes along the Alzheimer’s continuum, including a ‘biphasic’ behavior in which transient changes at the earliest disease stage are directionally counter to those associated with frank neurodegeneration at later disease stages. Novel cortical diffusivity parameters may be more sensitive than traditional metrics including standard mean diffusivity, as presented here, and offer a biologically-founded interpretation of microstructural changes in terms of cortical minicolumns, consistent with sequential phases of cytoarchitectural change as cortical ageing gives way to neuropathology. These imaging markers may help contribute to a more sensitive detection of the early stages of disease, and in the prognosis of subsequent macroscopic atrophy and cognitive decline.
Limitations
The combination of clinical, structural and diffusion MRI, amyloid PET and CSF data is a strength of this study, but it also creates a limitation in that the number of subjects with data for these disparate modalities is relatively small compared to the overall size of the ADNI study. Replication in larger cohorts, ideally with longitudinal data, will be necessary to confirm and expand the current findings.
Secondly, the participants were stratified or included in the study based on their amyloid status (negative and positive for cognitively normal individuals; only positive for patient groups) but the impact of tau in the studied cohort has not been explored. Although CSF pTau was considered in a secondary partial correlation analysis, a more comprehensive exploration of both fluid and PET measurement of tau (which appear to differ from each other in terms of timing and correlates (49)) warrants further study.
*Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
Funding: Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Acknowledgements: We would like to thank Dimitra Tzaferou for performing quality assessment of processing results.
Conflict of Interest Disclosure: SAC has submitted a patent application (WO2016162682A1) related to the diffusion MRI analysis used in the present study. SAC is a co-founder of a company, Oxford Brain Diagnostics, from which he has received funding for the research and preparation of this manuscript. MT and GRR are currently employed at a company, Oxford Brain Diagnostics. AJS was a full-time employee and shareholder of Takeda Pharmaceuticals, Ltd., when this work was performed.
Author Contributions: MT: conception and design of the study, analysis of data, drafting a significant portion of the manuscript and figures. GRR: analysis of data, drafting a significant portion of the manuscript. IH: analysis of data. AJS: conception and design of the study, drafting a significant portion of the manuscript. SAC: conception and design of the study, drafting a significant portion of the manuscript.
Ethical standards: The ADNI protocols were approved by all the Institutional Review Boards of the participating institutions. Only data from volunteers who had provided written informed consent were used to complete these analyses.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. Rakic P. A small step for the cell, a giant leap for mankind: A hypothesis of neocortical expansion during evolution. Trends in Neurosciences 1995;18:383-388 DOI: 10.1016/0166-2236(95)93934-p
2. Chance SA, Casanova MF, Switala AE, et al. Minicolumn thinning in temporal lobe association cortex but not primary auditory cortex in normal human ageing. Acta Neuropathol. 2006;111:459-64 DOI: 10.1007/s00401-005-0014-z
3. Chance SA, Clover L, Cousijn H, et al. Microanatomical correlates of cognitive ability and decline: normal ageing, MCI, and Alzheimer’s disease. Cerebral Cortex 2011;21(8):1870-1878 DOI: 10.1093/cercor/bhq264
4. van Veluw SJ, Sawyer EK, Clover L, et al. Prefrontal cortex cytoarchitecture in normal aging and Alzheimer’s disease: A relationship with IQ. Brain Structure and Function 2012;217(4):797-808 DOI: 10.1007/s00429-012-0381-x
5. Cruz L, Roe DL, Urbanc B, et al. Age-related reduction in microcolumnar structure correlates with cognitive decline in ventral but not dorsal area 46 of the rhesus monkey. Neuroscience 2009;158(4):1509-1520 DOI: 10.1016/j.neuroscience.2008.11.033
6. Buldyrev SV, Cruz L, Gomez-Isla T, et al. Description of microcolumnar ensembles in association cortex and their disruption in Alzheimer and Lewy body dementias. Proceedings of the National Academy of Sciences 2000;97(10):5039-5043 DOI: 10.1073/pnas.060009897
7. Whitwell JL, Avula R, Senjem ML, et al. Gray and white matter water diffusion in the syndromic variants of frontotemporal dementia. Neurology 2010;74(16):1279-1287 DOI: 10.1212/WNL.0b013e3181d9edde
8. Montal V, Vilaplana E, Alcolea D, et al. Cortical microstructural changes along the Alzheimer’s disease continuum. Alzheimer’s & Dementia 2018;14(3):340-351 DOI: 10.1016/j.jalz.2017.09.013
9. Montal V, Vilaplana E, Pegueroles J, et al. Biphasic cortical macro-and microstructural changes in autosomal dominant Alzheimer’s disease. Alzheimer’s & Dementia 2021;17(4): 618-628 DOI: 10.1002/alz.12224
10. Zhang H, Schneider T, Wheeler-Kingshott CA, & Alexander DC. NODDI: practical in vivo neurite orientation dispersion and density imaging of the human brain. Neuroimage 2012;61(4):1000-1016 DOI: 10.1016/j.neuroimage.2012.03.072
11. Parker TD, Slattery CF, Zhang J,et al.. Cortical microstructure in young onset Alzheimer’s disease using neurite orientation dispersion and density imaging. Human brain mapping 2018;39(7):3005-3017 DOI: 10.1002/hbm.24056
12. Wang Y, Wang Q, Haldar JP, et al. Quantification of increased cellularity during inflammatory demyelination. Brain 2011;134(12):3590-3601 DOI: 10.1093/brain/awr307
13. Torso M, Bozzali M, Zamboni G, et al.. Detection of Alzheimer’s Disease using cortical diffusion tensor imaging. Human Brain Mapping 2020;42.4: 967-977 DOI: 10.1002/hbm.25271
14. Torso M, Ahmed S, Butler C,et al. Cortical diffusivity investigation in posterior cortical atrophy and typical Alzheimer’s disease. Journal of Neurology 2020;268(1):227-239 DOI: 10.1007/s00415-020-10109-w
15. Torso M, Bozzali M, Cercignani M, et al. Using diffusion tensor imaging to detect cortical changes in fronto-temporal dementia subtypes. Scientific reports 2020;10(1):1-11 DOI: 10.1038/s41598-020-68118-8
16. Torso M, Ridgway GR, Jenkinson M, & Chance S. Intracortical diffusion tensor imaging signature of microstructural changes in frontotemporal lobar degeneration. Alzheimer’s Research & Therapy 2021;13(1):1-15 DOI: 10.1186/s13195-021-00914-4
17. McKavanagh R, Torso M, Jenkinson M,et al. Relating diffusion tensor imaging measurements to microstructural quantities in the cerebral cortex in multiple sclerosis. Human brain mapping 2019;40(15):4417-4431 DOI: 10.1002/hbm.24711
18. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institutes on Aging and the Alzheimer’s Association workgroup. Alzheimer’s & dementia 2011;7(3):263-269 DOI: 10.1016/j.jalz.2011.03.005
19. Fortin JP, Parker D, Tunç B, et al. Harmonization of multi-site diffusion tensor imaging data. Neuroimage 2017;161:149-170 DOI: 10.1016/j.neuroimage.2017.08.047
20. Kleinberger G, Yamanishi Y, Suárez-Calvet M, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Science translational medicine 2014;6(243):243ra86-243ra86 DOI: 10.1126/scitranslmed.3009093
21. Suarez-Calvet M, Caballero MA, Kleinberger G, et al. Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci Transl Med 2016;8 DOI: 10.1126/scitranslmed.aag1767
22. Suárez-Calvet M, Kleinberger G, Araque Caballero MÁ, et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol Med. 2016;8:466–76. Int. J. Geriatr. Psychiatry, 19, 817-824 DOI: 10.15252/emmm.201506123
23. Capell A, Liebscher S, Fellerer K, et al. Rescue of progranulin deficiency associated with frontotemporal lobar degeneration by alkalizing reagents and inhibition of vacuolar ATPase. Journal of Neuroscience 2011;31(5):1885-1894 DOI: 10.1523/JNEUROSCI.5757-10.2011
24. Benjamini Y, & Yekutieli D. The control of the false discovery rate in multiple testing under dependency. The Annals of Statistics 2001;29(4)
25. Ising C, Venegas C, Zhang S, et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019;575:669–673 DOI: 10.1038/s41586-019-1769-z
26. Zilka N, Kazmerova Z, Jadhav S, et al. Who fans the flames of Alzheimer’s disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J Neuroinflammation 2012;9:47 DOI: 10.1186/1742-2094-9-47
27. Sri S, Pegasiou CM, Cave CA, et al. Emergence of synaptic and cognitive impairment in a mature-onset APP mouse model of Alzheimer’s disease. Acta neuropathol commun 2019;7:25 DOI: 10.1186/s40478-019-0670-1
28. Akiyama H, Arai T, Kondo H,et al. Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis Assoc Disord. 2000;14 Suppl 1:S47-53 DOI: 10.1097/00002093-200000001-00008
29. Vogels T, Murgoci AN, & Hromádka T. Intersection of pathological tau and microglia at the synapse. Acta neuropathol commun 2019;7:109 DOI: 10.1186/s40478-019-0754-y
30. Selkoe DJ, & Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO molecular medicine 2016;8(6):595-608 DOI: 10.15252/emmm.201606210
31. Serrano-Pozo A, Frosch MP, Masliah E, & Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harbor perspectives in medicine 2011;1(1): a006189. DOI: 10.1101/cshperspect.a006189
32. Ulland TK, Song WM, Huang SCC, et al. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 2017;170(4):649-663 DOI: 10.1016/j.cell.2017.07.023
33. Mendsaikhan A, Tooyama I, & Walker DG. Microglial progranulin: involvement in Alzheimer’s disease and neurodegenerative diseases. Cells 2019;8(3):230. DOI: 10.3390/cells8030230
34. Wang Y, Cella M, Mallinson KJ, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015;160:1061-1071 DOI: 10.1016/j.cell.2015.01.049
35. Yeh FL, Wang Y, Tom I, et al. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 2016;91(2):328-340 DOI: 10.1016/j.neuron.2016.06.015
36. Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med 2013;368:117-127 DOI: 10.1056/NEJMoa1211851
37. Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat Commun. 2015;6:6176 DOI: 10.1038/ncomms7176
38. Huang Y, Happonen KE, Burrola PG, et al. Microglia use TAM receptors to detect and engulf amyloid β plaques. Nature Immunology 20221;22(5):586-594 DOI: 10.1038/s41590-021-00913-5
39. Yuan P, Condello C, Keene CD, et al. TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 2016;90(4):724-739 DOI: 10.1016/j.neuron.2016.05.003
40. Ma LZ, Tan L, Bi YL, et al. Dynamic changes of CSF sTREM2 in preclinical Alzheimer’s disease: the CABLE study. Molecular neurodegeneration 2020;15(1):1-9 DOI: 10.1186/s13024-020-00374-8
41. Edwin TH, Henjum K, Nilsson LN, et al. A high cerebrospinal fluid soluble TREM2 level is associated with slow clinical progression of Alzheimer’s disease. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring 2020;12(1):e12128 DOI: 10.1002/dad2.12128
42. Pereson S, Wils H, Kleinberger G, et al. Progranulin expression correlates with dense-core amyloid plaque burden in Alzheimer disease mouse models. J Pathol 2009;219: 173–181 DOI: 10.1002/path.2580
43. Minami SS, Min SW, Krabbe G, et al. Progranulin protects against amyloid β deposition and toxicity in Alzheimer’s disease mouse models. Nat Med 2014;20:1157-1164 DOI: 10.1038/nm.3672
44. Suárez-Calvet M, Capell A, Araque Caballero MÁ,et al. CSF progranulin increases in the course of Alzheimer’s disease and is associated with sTREM 2, neurodegeneration and cognitive decline. EMBO molecular medicine 2018;10(12):e9712 DOI: 10.15252/emmm.201809712
45. Wang Y, Ulland TK, Ulrich JD, et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016;213(5):667-675 DOI: 10.1084/jem.20151948
46. Piccio L, Deming Y, Del-Aguila JL, et al. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016;131:925-933 DOI: 10.1007/s00401-016-1533-5
47. Takahashi H, Klein ZA, Bhagat SM, et al. Opposing effects of progranulin deficiency on amyloid and tau pathologies via microglial TYROBP network. Acta neuropathologica 2017; 133(5):785-807 DOI: 10.1007/s00401-017-1668-z
48. Vilaplana E, Rodriguez-Vieitez E, Ferreira D, et al. Cortical microstructural correlates of astrocytosis in autosomal-dominant Alzheimer disease. Neurology 2020;94(19):e2026-e2036 DOI: 10.1212/WNL.0000000000009405
49. Ossenkoppele R, Reimand J, Smith R, Leuzy A, et al. Tau PET correlates with different Alzheimer’s disease-related features compared to CSF and plasma p-tau biomarkers. EMBO Mol Med. 2021;9;13(8):e14398. DOI: 10.15252/emmm.202114398.