
J. Delrieu1, R.J. Bateman2,3, J. Touchon4, M. Sabbagh5, J. Cummings6
CTAD Task Force members: Susan Abushakra (Framingham, USA); Paul Aisen (San Diego, USA); Sandrine Andrieu (Toulouse, France); Matthew Barton (Raleigh, USA); Monika Baudler (Basel, Switzerland); Joanne Bell (Willmington, USA); Tobias Bittner (Basel, Switzerland); Adam Boxer (San Francisco, USA) ; Dawn Brooks (Indianapolis, USA); Mirek Brys (Indianapolis, USA); Szofia Bullain (South San Francisco, USA); Cherie Butts (Cambridge, USA); Maria Carrillo (Chicago, USA); Carmen Castrillo-Viguera (Cambridge, USA); Bill Chan (Beijing, China); Ivan Cheung (Woodcliff Lake, USA); Min Cho (Woodcliff Lake, USA); Suzanne Craft (Winston-Salem, USA); Michael Detke (San Francisco, USA); Shobha Dhadda (Woodcliff Lake, USA); Rachelle Doody (Basel, Switzerland); Sanjay Dube (Viejo, USA); Billy Dunn (Beltsville, USA); Michael Egan (North Wales, USA); Rianne Esquivel (Malvern, USA); Colin Ewen (United Kingdom); Phyllis Ferrel (Indianapolis, USA); Michela Gallagher (Baltimore, USA); Wendy Galpern (New Jersey, USA); Hideki Garren (San Francisco, USA); Grönblad Anna-Kaija (Stockholm, Sweden); Juergen Haeussler (Titusville, USA); Harald Hampel (Woodcliff Lake, USA); Suzanne Hendrix (Salt Lake City, USA); Joseph Herring (North Wales, USA); Michael Irizarry (Woodcliff Lake, USA); Takeshi Iwatsubo (Tokyo, Japan); Gene Kinney (San Francisco, USA); David Knopman (Rochester, USA); Hartmuth Kolb (Titusville, USA); Shailaja Korukonda (Woodcliff Lake, USA); Akihiko Koyama (Woodcliff Lake, USA); Lynn Kramer (Woodcliff Lake, USA); Luka Kulic (Basel, Switzerland); Ricky Kurzman (Woodcliff Lake, USA); Jaren Landen (Cambridge, USA); Lars Lannfelt (Uppsala, Sweden); John Lawson (Malvern, USA); Valérie Legrand (Nanterre, France); Jinhe Li (Gilbert, USA); Frank Longo (Stanford, USA); Manoj Malhotra (Woodcliff Lake, USA); William Menard (Providence, USA); Mark Mintun (Indianapolis, USA); Cecilia Monteiro (South San Francisco, USA); Stacie O’Sullivan (Woodcliff Lake, USA); Tomas Odergren (Stockholm, Sweden); Gunilla Osswald (Stockholm, Sweden); Ronald Petersen (Rochester, USA); Michael Pontecorvo (Indianapolis, USA); Mary Ellen Quiceno (New Jersey, USA); Rema Raman (San Diego, USA); Larisa Reyderman (Woodcliff Lake, USA); Monica Rivera-Mindt (Bronx, USA); Sharon Rogers (Los Angeles, USA); Sharon Rosenzweig-Lipson (Baltimore, USA); Ivana Rubino (Cambridge, USA); Laurie Ryan (Bethesda, USA); Stephen Salloway (Providence, USA); Rachel Schindler (New York, USA); Lon Schneider (Los Angeles, USA); Peter Schüler (Langen, Germany); Hiroshi Sekiya (Malvern, USA); Dennis Selkoe (Boston, USA); Melanie Shulman (Cambridge, USA); Eric Siemers ((Zionsville, USA); John Sims (Indianapolis, USA) Kaycee Sink (South San Francisco, USA); Reisa Sperling (Boston, USA); Joyce Suhy (San Mateo, USA); Chad Swanson (Woodcliff Lake, USA); Jina Swartz (London, United Kingdom); Pierre Tariot (Phoenix, USA); Edmond Teng (South San Francisco, USA); Martin Traber (Basel, Switzerland); Dominic Walsh (Cambridge, USA); Michael Weiner (San Francisco, USA); Lisa Yarenis (Woodcliff Lake, USA); Wagner Zago (San Francisco, USA); Kenton Zavitz (Cambridge, United Kingdom).
1. Maintain Aging Research team, CERPOP, Université de Toulouse, Inserm, Université Paul Sabatier, Toulouse, France; Gérontopôle, Department of Geriatrics, Toulouse CHU, Toulouse, France; 2. Department of Neurology, Washington University School of Medicine, St. Louis, MO, USA; 3. Dominantly Inherited Alzheimer Network Trials Unit, Washington University School of Medicine, St. Louis, MO, USA; 4. University of Montpellier, Montpellier, France; 5. Barrow Neurological Institute, Phoenix, Arizona, USA; 6. Chambers-Grundy Center for Transformative Neuroscience, Department of Brain Health, School of Integrated Health Sciences, University of Nevada Las Vegas., Las Vegas, NV 89154, USA.
Corresponding Author: Julien Delrieu, Pôle gériatrie, Cité de la santé, Place Lange – TSA 60033 – 31059 Toulouse Cedex 9, INSERM UMR 1027, faculté de médecine, 37 allées Jules Guesde. 31000 Toulouse, delrieu.j@chu-toulouse.fr
J Prev Alz Dis 2022;3(9):393-399
Published online May 16, 2022, http://dx.doi.org/10.14283/jpad.2022.48
Abstract
BACKGROUND: Aducanumab (ADUHELMTM) was approved for the treatment of Alzheimer’s disease (AD) in the US. This approval was supported by an effect on the cerebral amyloid plaque load and evidence of cognitive efficacy to be confirmed in post-marketing trials. Other anti-amyloid antibodies are under investigation in phase III (donanemab, lecanemab, gantenerumab) and have shown preliminary evidence of a cognitive benefit in phase II trials. Although these agents target a small segment of patients with mild cognitive impairment due to AD or mild AD dementia, their advent will change the design of future clinical trials both for anti-amyloid and non-amyloid drugs. These changes will promote the selection of patients in clinical trials by amyloid and tau biomarkers that identify patients with appropriate biology and may follow the treatment response to approved amyloid antibodies. The use of these agents creates the opportunity to test combined drug therapies and to conduct comparative assessments with innovative therapies and newly approved drugs available in clinical practice. Blood-based AD biomarkers should be implemented in research and could facilitate the recruitment into clinical trials. Anti-amyloid antibodies will have positive (e.g., more early diagnosis) and negative impacts (some subjects will be reluctant to participate in trials and risk assignment to placebo) on AD trials in the immediate future. We present the results of the CTAD Task Force on this topic, in Boston, November 6, 2021.
Key words: Alzheimer disease, clinical trial, anti-amyloid therapy, biomarker.
Introduction
Recently, aducanumab (ADUHELMTM) was approved for the treatment of Alzheimer’s disease (AD) in the United States (US). This anti-amyloid monoclonal antibody demonstrated an effect on the cerebral amyloid plaque burden and showed preliminary evidence of efficacy (in the PRIME phase Ib trial and the EMERGE but not ENGAGE phase III trials) in patients who had received the high doses (1–4). Numerous other phase III trials with anti-amyloid antibodies are underway and will potentially add to the repertoire of anti-amyloid therapies for the treatment of AD. Gantenerumab is currently being studied in the GRADUATE program (NCT03444870) and the GRADUATION trial (NTC04592341), donanemab in the TRAILBLAZER-ALZ 2, 3, 4 and EXT trials (NCT04640077, NCT04437511, NCT05026866 and NCT05108922) and lecanemab in the CLARITY (NCT03887455) and AHEAD 3-45 (NCT04468659) trials. These therapies could markedly change the therapeutic landscape of AD.
Anti-amyloid antibodies may have both positive and negative impacts on AD trials in the immediate future (5). This includes potential diversion of resources from clinical research to support clinical care including staff time and space. It may impact enrollment and recruitment if patients need to choose between an approved treatment or a clinical trial/observational study. The approval of ADUHELMTM and the potential arrival of other anti-amyloid antibodies for clinical practice may change the design of future therapeutic trials both for anti-amyloid and non-amyloid therapies. To date, the approval of ADUHELMTM beyond the US is uncertain (refusal recommended by Europe Medicines Agency but ongoing re-examination, request for more data by the Japanese Health Ministry) and some of the considerations discussed in this paper may not be applicable globally (6). Furthermore, using clinical trials criteria, estimates of eligible subjects for aducanumab are low and range from 8-20% of patients with AD (7).
The EU/US CTAD Task Force met in November 2021 at Boston to discuss this topic, bringing together a global group of clinical investigators from academia and industry. The CTAD Task Force assessed the consequences of advent of anti-amyloid therapies for amyloid and non-amyloid trial design:
1) What issues need to be addressed in future trials of ADUHELMTM?
2) What are the consequences of ADUHELMTM approval for inclusion criteria in future clinical trials?
3) What is the place for biomarkers in labeling of new agents and for blood-based biomarkers in future AD clinical trials?
4) What are consequences in prevention trials such as AHEAD 3-45 and DIAN-TU NextGen?
Aducanumab: prescribing instructions, appropriate use recommendations and issues to be resolved
Label prescribing instructions
ADUHELMTM is a human anti-amyloid monoclonal antibody indicated for the treatment of AD (3). According the Food and Drug Administration (FDA) updated label, ADUHELMTM can be prescribed for patients with mild cognitive impairment (MCI) or mild dementia due to AD (4). The cognitive benefit will require verification in confirmatory post-marketing trials to maintain this indication. This indication was approved using an accelerated approval regulatory pathway supported by removal of amyloid plaques observed in phase Ib and III trials. EMERGE and ENGAGE trials showed that higher exposures to ADUHELMTM were associated with greater effect on cognitive decline and cerebral amyloid plaque load. The reduction of amyloid plaques was demonstrated on amyloid positron emission tomography (PET). An association between reduction in cerebral amyloid burden and cognitive decline (Clinical Dementia Rating-sum of boxes [CDR-SB] as primary outcome) was observed. In sub-studies, ADUHELMTM with high doses showed an impact on tau pathology both by decreasing cerebrospinal fluid (CSF) levels of p-tau and t-tau (in the EMERGE study [n=17] but not significantly in the ENGAGE study [n=18]), and brain tau pathology assessed by tau PET in the medial temporal, temporal, and frontal but not in parietal and cingulate regions (pooled analysis, n=37). As mentioned previously, the effects of ADUHELMTM on AD biomarkers is described in the label but the need for a positive amyloid biomarker to qualify for therapy is not explicitly required in the updated label.
For the monitoring of amyloid related imaging abnormalities (ARIAs), the US FDA Prescribing Instructions recommend obtaining recent (within one year) brain magnetic resonance imaging (MRI) before initiating therapy and MRIs before the 7th infusion (first high dose) and 12th infusion (sixth high dose). The treatment may be continued with caution if new microhemorrhages of focal superficial siderosis are observed. The label prescribing instructions are different from the criteria for inclusion in EMERGE and ENGAGE trials creating uncertainty and potentially posing a problem regarding the safety of ADUHELMTM in clinical practice.
Appropriate use recommendations and issues in clinical practice
Currently, many subjects with a family history of AD consult physicians at an early stage of symptoms. If the drug is available, both patients and physicians may change their behavior and practice and this trend will probably increase (8). An expert panel developed appropriate use recommendations to help clinicians translate the Prescribing Instructions and clinical trial data into patient care (9).
Several studies have shown that the inclusion and exclusion criteria from EMERGE and ENGAGE studies are probably too restrictive and selected patients not representative of “real world” populations. For patients enrolled in Medicare, 91.0% with AD and 85.5% with MCI met at least 1 trial exclusion criterion. The most common exclusion criteria identified were chronic kidney and cardiovascular diseases, anticoagulation, and advanced age (over 85 years) (7). In a geriatric environment of a European university hospital (Italy), the results of another study are similar (10). As judged by the clinicians, patients were ineligible for ADUHELMTM due to age, low education level, absence of a caregiver, cognitive impairment too severe, compromised autonomy, a major laboratory abnormality or/and a significant brain vascular disease. Thus, only a very low proportion of patients (<1%, potentially underestimated) with cognitive disorders would be potentially eligible from ADUHELMTM in clinical practice. The expert panel made multiple recommendations that expand the population beyond the trial population.
In contrast to the clinical trials criteria, the label Prescribing Instructions are broad. The expert panel recommendations suggest that the appropriate use of ADUHELMTM in practice should replicate the use of aducanumab in the phase III trials in particular the confirmation of a positive amyloid status prior to prescription (9). The indications would be more limited and safety recommendations more restrictive than the information provided in the label Prescribing Instructions (9). MRI monitoring may be needed more often and earlier than suggested by FDA, particularly in APOE4 carriers.
Safety issues to be addressed in post-marketing trials
Many issues need to be addressed in future trials with anti-amyloid therapies: ARIA monitoring and management, special circumstances (autosomal dominant AD, atypical forms, Down syndrome with cognitive decline, and others), and indications and contraindications in the “real-world”. One major concern to address is the growing number of older persons taking anti-coagulant medications who have been excluded from clinical trials and considered also as contraindication by expert panel recommendations. Impaired clotting may increase the risk of making ARIA-H (ARIA-Hemorrhage) side-effects more significant. Post-marketing trials with aducanumab should clarify these points to optimize use of anti-amyloid antibodies in practice. Three trials are planned by the sponsor to complete phase III data: 1) a re-dosing phase III study (EMBARK, NCT04241068), 2) a phase IV confirmatory study, and 3) an observational Phase IV 5-year study called ICARE-AD (NCT05097131), introduced at the Alzheimer’s Association International Conference 2021 (Amsterdam), to assess real-world effectiveness and safety. The advent of aducanumab will also allow comparison studies such as the TRAILBLAZER ALZ IV study (NCT05108922) which will compare the effects of donanemab and aducanumab on brain amyloid plaque load.
What are the potential consequences for inclusion criteria and sample size in future clinical trials?
Removal of amyloid will have consequences for future trial design including participation criteria. The AD drug development pipeline has many classes of drugs affecting different targets (11). However, even non-amyloid target trials include subjects based on amyloid biomarkers (CSF or PET and perhaps blood-based markers in the future). High doses of amyloid antibodies can change the amyloid status of participants and this effect seems to be cumulative over time (see table 1). In this context, how will recruitment into new AD clinical trials work if patients are taking anti-amyloid treatments and are potentially missing this characteristic AD marker? Patients whose amyloid plaque burden has been reduced to undetectable levels no longer meet amyloid-tau-neurodegeneration (AT[N]) criteria for AD (12). A solution to consider could be the use of tau biomarkers as inclusion criteria in future trials. Currently, the selection of subjects based on tau biomarkers in clinical trials is difficult. In the phase II TRAILBLAZER-ALZ trial, selection by biomarker clearly affected screening and enrollment (screen failure of 30% due to tau-PET status) (13). Anticipating that anti-amyloid antibodies can influence tau pathology level and soluble tau biomarkers; patient selection is likely to be even more difficult in the future.
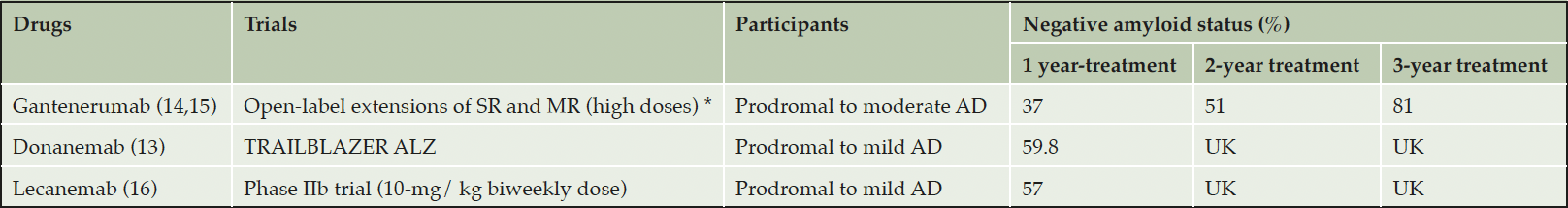
Table 1. Examples of amyloid antibodies effect on binary amyloid status assessed by PET
SR, SCarlet RoAD; MR, Marguerite RoAD; AD, Alzheimer Disease; UK, UnKnown; *15% of participants had a negative amyloid status at baseline visit of open-label extensions.
By acting on amyloid pathology, antibodies also affect a multitude of other processes involved in AD including tau pathology and soluble tau biomarkers. This was demonstrated in clinical trials (see table 2) and is evident in data from the Alzheimer Disease Neuroimaging Initiative (ADNI) (17). This extensive evidence evokes a close relationship between amyloid and tau; tau pathology spread accelerates once amyloid load reaches a critical cut-off launching a cascade of metabolic, degenerative, and cognitive changes. Tau-PET studies highlighted a temporal and spatial pattern of tau pathology dependent on prior Aβ deposition and related to subsequent cognitive decline.
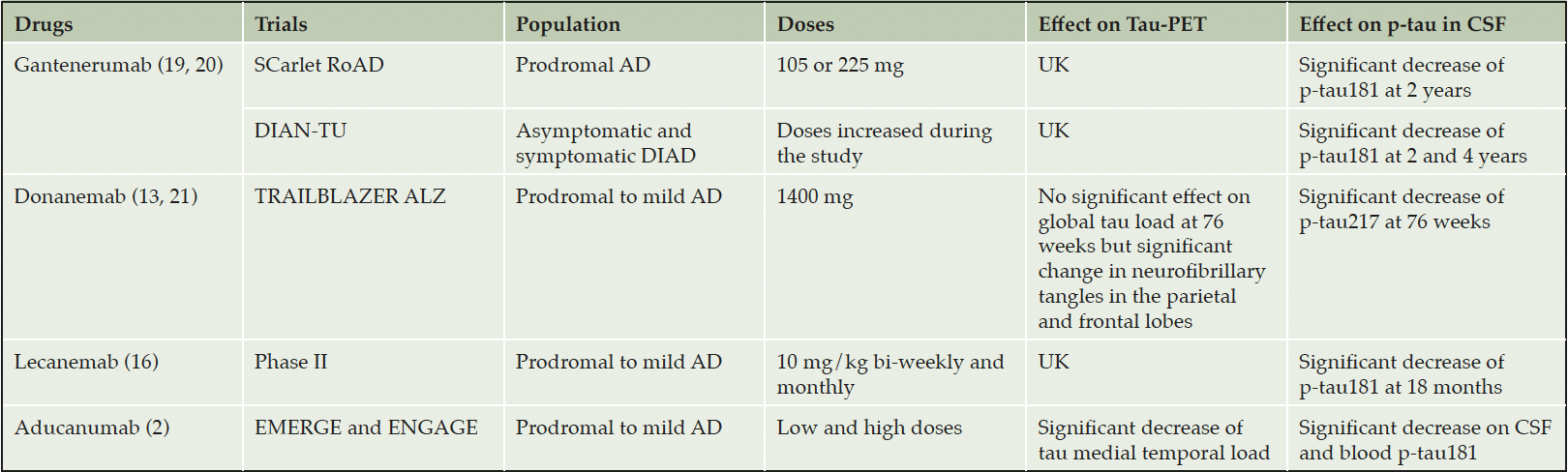
Table 2. Effect of amyloid antibodies on tau biomarkers assessed by PET and CSF measures
DIAN-TU, Dominantly Inherited Alzheimer Network Trial Unit; DIAD, Dominantly Inherited Alzheimer’s Disease; AD, Alzheimer Disease; UK, UnKnown.
It is important to consider that decreasing amyloid pathology will change this cascade of events and this should be considered in the recruitment of future clinical trials including sample size and power calculation. Amyloid and tau effects will slow disease progression and achieving a 0.5 drug-placebo difference on the CDR-SB will likely require a larger sample size in future clinical trials. Power calculation estimates might increase sample size by 30-50% to detect drug-placebo differences in a population that progresses more slowly than untreated AD. Alternatively, removing amyloid plaques may have an additive or synergistic effect on tau drug efficacy, by removing the amyloid mechanisms that drive tau spread pathophysiology including tau over-production due to amyloid plaques (18). Anti-tau drugs may be more effective in a population that has received anti-amyloid treatment, but this may require a much longer trial to demonstrate.
Place of blood-based and non-blood-based biomarkers in AD trials
How to include biomarkers in the product label?
As mentioned above, ADUHELMTM is indicated in patients with MCI or mild dementia stage of AD (4). This indication is not based on biomarkers on the Prescribing Instructions whereas its approval is supported mainly by removal of amyloid plaque load. Biomarkers are mentioned only in the label section 14 for detailing the impact of ADUHELMTM on amyloid and tau pathologies. In 2021, more than 40% of phase II and III trials used biomarkers including amyloid PET or CSF as inclusion criteria (22). This point raises the question of how AD biomarkers should be mentioned in the label in the field of AD, especially for the indication(s). We have many examples of drugs whose indication is based on biomarkers in the cardiovascular field:
– The first indication of LIPITOR (atorvatin) approved in 1999 is clinical and second indication is based on biomarker (reduce elevated total-C, LDL-C, apo B, and TG levels and increase HDL-C in patients with primary hyperlipidemia).
– CRESTOR (rosuvastatin) has been approved in 2003, all indications are based on biomarkers.
– TRUCILITY (dulaglutide) is indicated as “an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus”. The second indication is clinical.
With accelerated approvals, a biomarker should be “reasonably likely” to predict clinical benefit. The reduction of amyloid plaque load is associated with the slowing of cognitive decline in several studies (2, 13, 16, 23). However, the clinical benefit of anti-amyloid antibodies may be mediated by secondary mechanisms beyond removal of amyloid pathology including impact on tau toxicity, neuro-inflammation, or neurodegeneration. These secondary affects need to be accommodated in product labeling (if relevant data of sufficient quality are generated) as well as in future trial design including non-amyloid trials. More studies are needed to confirm and use amyloid plaque load as a surrogate marker similar to the use of glycemic or cholesterol levels in the cardiovascular field. Other potential surrogate biomarkers include tau PET, soluble amyloid-beta42/40, and soluble forms of tau. If confirmed as a predictive surrogate outcome, amyloid plaque burden or another biomarker, could be utilized as a primary endpoint could allow for shorter future trials with fewer participants and thus accelerate the discovery of new therapies. As information evolves, biomarkers (plasma or otherwise) that define inclusion criteria and that may reflect efficacy could be included in labels.
Place of blood-based biomarkers for future AD trials?
To date, amyloid level assessed by PET or CSF measures are the most widely used biomarkers to select participants for clinical trials of AD disease-modifying therapies. However, screening by amyloid PET is difficult to generalize in clinical practice given its cost and limited access (in Europe and much of the rest of the world). Thus, the use of innovative therapies may be difficult in clinical practice, especially if biomarkers are included in the label of new treatments in the future. In the Multidomain Alzheimer Prevention Trial (MAPT), low plasma Aβ42/40 was associated with an increased cognitive decline in non-demented participants (MCI and cognitively unimpaired subjects) over time (24). In the Biomarkers For Identifying Neurodegenerative Disorders Early and Reliably (BioFINDER) study, blood-based biomarkers (Aβ42/40 ratio, p-tau217 and neurofilament light chain, NfL) predict cognitive decline and incident AD dementia in cognitively unimpaired subjects (25). In the BioFINDER and ADNI studies, a model combining blood p-tau, memory with executive tests, and apolipoprotein E (APOE) genotype predicts the risk of developing dementia in subjects with subjective cognitive decline and MCI (area under the curve = 0.90-0.91) (26). Using CSF biomarkers instead of blood-based biomarkers did not improve the prediction accuracy of this model. The presence of amyloid plaques was detected with similar factors across ADNI, BioFINDER, and Australian, Imaging, Biomarkers and Lifestyle (AIBL) cohorts (27). These findings suggest that blood-based biomarkers (Aβ42/40 ratio or/and p-tau) may be used to identify in non-demented subjects the risk of cognitive decline and for developing AD dementia. The use of blood-based biomarkers could certainly facilitate recruitment with more cost and time effective screening or prescreening by reducing the number of patients that need to go on to more advanced diagnostics prior to trial qualification or exclusion. Exploratory economic analyses from the ADNI study showed that the use of blood amyloid biomarkers as a prescreening tool prior to amyloid PET significantly reduced screening cost expected in an AD prevention trial (28). The deployment of blood-based biomarkers could also simplify similarly the use of innovative therapies in clinical practice in the future.
The example of DIAN-TU program
DIAN-TU NextGen: background and objectives
The Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU) study observed no cognitive impact of lower dose gantenerumab and solanezumab in symptomatic and asymptomatic Dominantly inherited Alzheimer’s disease (DIAD). However, gantenerumab demonstrated an effect on AD biomarkers, decreasing cerebral amyloid load (Pittsburgh Compound-B [PIB]-PET), and measures of t-tau, p-tau181, and NfL in CSF (19). In view of its encouraging effects on biomarkers, an exploratory open-label extension with gantenerumab, testing of another anti-amyloid monoclonal antibody (lecanemab), and non-amyloid treatments are planned in the DIAN-TU NextGen program. Tau pathology is an attractive target among non-amyloid targets and tau may be playing a role in the early stages of DIAD. The early soluble tauopathy hyperphosphorylation (increase in p-tau217 and p-tau181) with the onset of fibrillar amyloid pathology seems to play a determining role in the asymptomatic stages of AD, whereas tau aggregation (assessed by tau-PET) is more involved in the symptomatic stages (29). The strongest association between cognitive performance and AD pathology is with neurofibrillary tangles, and markers of brain tau pathology may be good biomarkers to track and stage cognitive decline (better than soluble forms of p-tau181 or 217). These data are crucially important and, if these hypotheses confirmed, should be taken into consideration in the design of future clinical trials evaluating anti-tau therapies as informed by DIAN-TU NextGen observations.
The advent of anti-amyloid therapies has also provided opportunities to launch combination trials, to provide potentially greater biologic and clinical benefit, to improve biological understanding of the relationship between amyloid and tau pathologies, and to better understand the drug impact of anti-amyloid antibodies on multiple tau and other downstream mechanisms. In this context, the specific aims of the DIAN-TU Tau NexGen are: 1) to demonstrate biological engagement of tau and/or combined drugs to significantly decrease tau aggregation measured by tau-PET, 2) to determine if neurodegeneration, hypometabolism, and inflammatory AD processes show fewer changes in the active treatment group compared to the control group and 3) to show potential slowing of cognitive decline to support the transition to phase III validation studies.
How to account for the advent of anti-amyloid therapies in DIAN-TU Tau NextGen design?
To take advantage of the advent of anti-amyloid antibody therapies, changes in the design of the trials are needed. The ability to test anti-tau monotherapy in future drug arms is likely limited, especially for symptomatic participants who may desire treatment with available anti-amyloid treatment. Allowing anti-amyloid therapies could be required for participant recruitment and retention. The segregation of participants across symptomatology (asymptomatic vs symptomatic) and/or tau pathology levels (negative vs positive status), the order of administration (treat tau first or amyloid first or at the same time?) and the duration of monotherapy should be considered in future designs. Given these design considerations, the DIAN-TU tau next generation platform has designed trials of anti-tau therapies in the following way (see Figure 1). In symptomatic patients, anti-amyloid drug would be administered first for 6 months, then adding an anti-tau drug (randomization 1:1). In asymptomatic subjects, an anti-tau drug would be administered first for 12 months (randomization 1:1), followed by the addition of an anti-amyloid drug. The use of biomarkers as surrogate endpoints and as inclusion criteria must be adapted to stage of the disease. In asymptomatic participants, CSF p-tau, t-tau and NfL could be used as endpoints and negative tau-PET or CDR 0 as inclusion criteria. In symptomatic patients, tau-PET and neurodegeneration biomarkers (FDG-PET, NfL, and brain atrophy) could be used as endpoints and positive tau-PET or CDR>0 as inclusion criteria. The first trial arm utilizing this design has launched with lecanemab and E2814, an anti-tau antibody (NCT #).
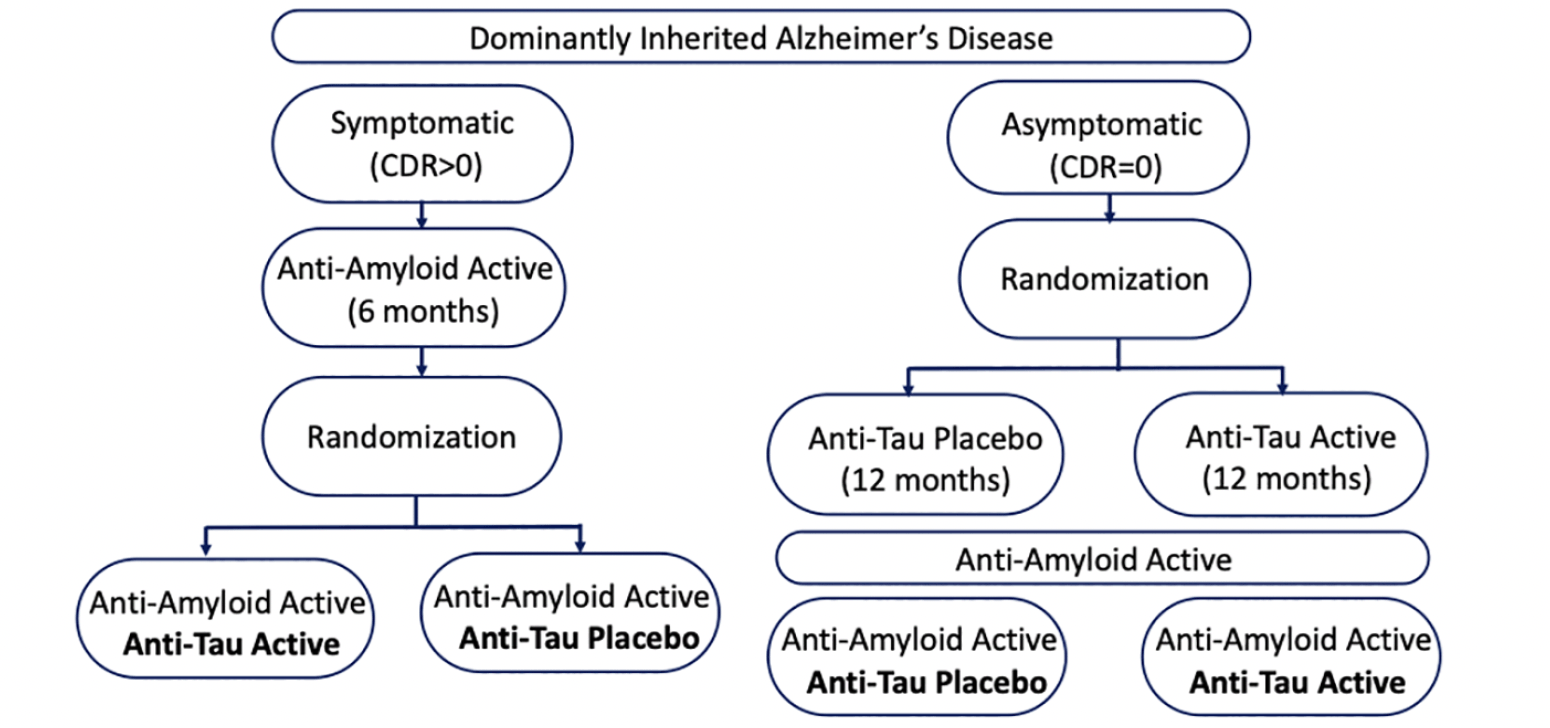
Figure 1. Anti-tau therapies in the DIAN-TU tau next generation platform
CDR, Clinical Dementia Rating.
Non-DIAN-TU Trial Design Considerations
The approval of ADUHELMTM must be accommodated in future trials of patients who meet appropriate use criteria. Several options are available. First, patients on ADUHELMTM might be excluded from trials of novel therapies as is currently done in some trials where the standard of care with donepezil or memantine is excluded. This must be considered from an ethical perspective since delay of symptomatic therapies may have different consequences from delay of a disease-modifying therapies (DMT). Second, patients may be allowed into trials irrespective of their treatment with other stable DMTs such as aducanumab with planned analyses based on the presence or absence of the DMT or recruitment may be stratified based on DMT status. This approach will have effects on expected effects size, power calculations, and sample sizes. Third, comparative studies could be conducted to determine the relative of clinical efficacy, biomarker efficacy, or safety of the agents in the trial. These could have superiority, non-inferiority, of mixed (e.g., non-inferior clinical efficacy and superior safety) designs. These approaches can be considered as the field matures and experience with DMTs increases.
Conclusion
The advent of anti-amyloid therapies in clinical practice will have both positive and negative effects on future clinical trials. These changes will not have impact world-wide at first, at present ADUHELMTM has received approval only in the US. Furthermore, anti-amyloid immunotherapies will target a small segment of the AD population. The arrival of new treatments will likely change the behavior of general practitioners (GPs), patients and their families. The lack of effective treatment is one of the main reasons for patients not to go for consultation and for GPs not to refer to a memory clinic. The availability of a treatment should encourage early diagnosis of AD. The negative effects of the approval of aducanumab are related to the potentially more difficult recruitment, the interest in future trials could be reduced in favor of an approved treatment. Removal of brain amyloid pathology by anti-amyloid antibodies will also have consequences in future trial design for selection of participants.
The advent of new treatments is also an opportunity to test the efficacy of combined therapies and to provide comparison trials with approved or emerging drugs. In particular, combining anti-tau therapies with the anti-amyloid effect of approved therapies is a compelling challenge and opportunity for future clinical trials. In future therapeutic trials, biomarkers can be used to better predict cognitive decline and treatment efficacy. Tau-PET appears to be a promising biomarker to predict the cognitive change in symptomatic stages of AD. Blood biomarkers could be implemented for selection and monitoring drug response in asymptomatic stages. Once validated as surrogate endpoints, these biomarkers should be described in the label indications and not only to report the non-clinical effects.
Conflict of interest: The Task Force was partially funded by registration fees from industrial participants. These corporations placed no restrictions on this work. Dr. Delrieu has received payment/honoraria from Biogen (presentation for Biogen in 2021); has participated on a Data Safety Monitoring Board or Advisory Board for French board for Roche in 2020-2022. Dr. Bateman is Director of DIAN–TU and Principal Investigator of DIAN–TU001. He receives research support from the NIA of the NIH, DIAN–TU trial pharmaceutical partners (Eli Lilly and Company, F. Hoffman La Roche Ltd, Janssen, Eisai, Biogen and Avid Radiopharmaceuticals), Alzheimer’s Association, GHR Foundation, Anonymous Organization, DIAN–TU Pharma Consortium (active: Biogen, Eisai, Eli Lilly and Company, Janssen, F. Hoffmann La Roche Ltd/Genentech; previous: AbbVie, Amgen, AstraZeneca, Forum, Mithridion, Novartis, Pfizer, Sanofi, United Neuroscience), NfL Consortium (Hoffman La-Roche, Biogen, AbbVie, and Bristol Meyer Squibb) and Tau SILK Consortium (Eli Lilly and Company, Biogen and AbbVie) . He has been an invited speaker for the Korean Dementia Association and American Neurological Association and a consultant for Amgen, Hoffman La-Roche and Eisai. D.H., is an inventor on patents for one of the treatments (solanezumab), which was tested in the DIAN clinical trials. If solanezumab is approved as a treatment for Alzheimer’s disease or Dominantly Inherited Alzheimer’s Disease, Washington University and Dr. Holtzman will receive part of the net sales of solanezumab from Eli Lilly, which has licensed the patents related to solanezumab from Washington University. Dr. N. Sabbagh: consulting fees (Alzheon, Neurotrope, Biogen, Cortexyme, Danone, Regeneron, Roche-Genentech, Stage 2 Innovations, Acadia); stock or stock options (Brain Health Inc, NeuroReserve, NeuroTau, Optimal Cognitive Health Company, uMethod Health,Versanum, Athira). Dr. Cummings has provided consultation to AB Science, Acadia, Alkahest, AlphaCognition, ALZPath, Annovis, AriBio, Artery, Avanir, Biogen, Cassava, Cerevel, Clinilabs, Cortexyme, Diadem, EIP Pharma, Eisai, GatehouseBio, GemVax, Genentech, Green Valley, Grifols, Janssen, Karuna, Lexeo, Lilly, Lundbeck, LSP, Merck, NervGen, Novo Nordisk, Oligomerix, Otsuka, PharmacotrophiX, PRODEO, Prothena, ReMYND, Renew, Resverlogix, Roche, Signant Health, Suven, Unlearn AI, Vaxxinity, VigilNeuro, Zai Laboratories pharmaceutical, assessment, and investment companies. He is supported by NIGMS grant P20GM109025; NINDS grant U01NS093334; NIA grant R01AG053798; NIA grant P20AG068053; NIA grant R35AG71476; Alzheimer’s Disease Drug Discovery Foundation (ADDF); and the Joy Chambers-Grundy Endowment.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016 01;537(7618):50–6.
2. Budd Haeberlein S, von Hehn C, Tian Y, et al. EMERGE and ENGAGE Topline Results: Two Phase 3 Studies to Evaluate Aducanumab in Patients With Early Alzheimer’s Disease. In: 12th Clinical Trials on Alzheimer’s Disease. San Diego, 2019.
3. U.S. Food & Drug Administration. Drugs@FDA: FDA-Approved Drugs. Aducanumab. Reference ID 4822820 2021.
4. U.S. Food & Drug Administration. Drugs@FDA: FDA-Approved Drugs — Aducanumab. Reference ID 4807032. 2021.
5. Weiner MW, Aisen PS, Beckett LA, Green RC, Jagust W, Morris JC, et al. Editorial: How Will Aducanumab Approval Impact AD Research? J Prev Alzheimers Dis. 2021;8(4):391–2.
6. Mahase E. Aducanumab: European agency rejects Alzheimer’s drug over efficacy and safety concerns. BMJ. 2021 Dec 20;375:n3127.
7. Anderson TS, Ayanian JZ, Souza J, Landon BE. Representativeness of Participants Eligible to Be Enrolled in Clinical Trials of Aducanumab for Alzheimer Disease Compared With Medicare Beneficiaries With Alzheimer Disease and Mild Cognitive Impairment. JAMA. 2021 Oct 26;326(16):1627–9.
8. Vellas BJ. Editorial: The Geriatrician, the Primary Care Physician, Aducanumab and the FDA Decision: From Frustration to New Hope. J Nutr Health Aging. 2021;25(7):821–3.
9. Cummings J, Aisen P, Apostolova LG, Atri A, Salloway S, Weiner M. Aducanumab: Appropriate Use Recommendations. J Prev Alzheimers Dis. 2021;8(4):398–410.
10. Canevelli M, Rossi PD, Astrone P, Consorti E, Vanacore N, Cesari M. “Real world” eligibility for aducanumab. J Am Geriatr Soc. 2021 Oct;69(10):2995–8.
11. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement (N Y). 2020;6(1):e12050.
12. Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14(4):535–62.
13. Mintun MA, Lo AC, Duggan Evans C, Wessels AM, Ardayfio PA, Andersen SW, et al. Donanemab in Early Alzheimer’s Disease. N Engl J Med. 2021 Mar 13;NEJMoa2100708.
14. Klein G, Delmar P, Voyle N, Rehal S, Hofmann C, Abi-Saab D, et al. Gantenerumab reduces amyloid-β plaques in patients with prodromal to moderate Alzheimer’s disease: a PET substudy interim analysis. Alzheimers Res Ther. 2019 Dec 12;11(1):101.
15. Klein G, Delmar P, Kerchner GA, Hofmann C, Abi-Saab D, Davis A, et al. Thirty-Six-Month Amyloid Positron Emission Tomography Results Show Continued Reduction in Amyloid Burden with Subcutaneous Gantenerumab. J Prev Alzheimers Dis. 2021;8(1):3–6.
16. Swanson CJ, Zhang Y, Dhadda S, Wang J, Kaplow J, Lai RYK, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res Ther. 2021 Apr 17;13(1):80.
17. Veitch DP, Weiner MW, Aisen PS, Beckett LA, Cairns NJ, Green RC, et al. Understanding disease progression and improving Alzheimer’s disease clinical trials: Recent highlights from the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement. 2019 Jan;15(1):106–52.
18. Sato C, Barthélemy NR, Mawuenyega KG, Patterson BW, Gordon BA, Jockel-Balsarotti J, et al. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron. 2018 Mar 21;97(6):1284-1298.e7.
19. Salloway S, Farlow M, McDade E, Clifford DB, Wang G, Llibre-Guerra JJ, et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat Med. 2021 Jul;27(7):1187–96.
20. Ostrowitzki S, Lasser RA, Dorflinger E, Scheltens P, Barkhof F, Nikolcheva T, et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther. 2017 Dec 8;9(1):95.
21. Symposia — Oral Communications — Late Breaking News. The Journal of Prevention of Alzheimer’s Disease. 2021 Nov 1;8(1):S1–72.
22. Cummings J, Lee G, Zhong K, Fonseca J, Taghva K. Alzheimer’s disease drug development pipeline: 2021. Alzheimers Dement (N Y). 2021;7(1):e12179.
23. Avgerinos KI, Ferrucci L, Kapogiannis D. Effects of monoclonal antibodies against amyloid-β on clinical and biomarker outcomes and adverse event risks: A systematic review and meta-analysis of phase III RCTs in Alzheimer’s disease. Ageing Res Rev. 2021 Jul;68:101339.
24. Giudici KV, de Souto Barreto P, Guyonnet S, Li Y, Bateman RJ, Vellas B, et al. Assessment of Plasma Amyloid-β42/40 and Cognitive Decline Among Community-Dwelling Older Adults. JAMA Netw Open. 2020 Dec 1;3(12):e2028634.
25. Cullen NC, Leuzy A, Janelidze S, Palmqvist S, Svenningsson AL, Stomrud E, et al. Plasma biomarkers of Alzheimer’s disease improve prediction of cognitive decline in cognitively unimpaired elderly populations. Nat Commun. 2021 Jun 11;12(1):3555.
26. Palmqvist S, Tideman P, Cullen N, Zetterberg H, Blennow K, Alzheimer’s Disease Neuroimaging Initiative, et al. Prediction of future Alzheimer’s disease dementia using plasma phospho-tau combined with other accessible measures. Nat Med. 2021 Jun;27(6):1034–42.
27. Li Y, Schindler SE, Bollinger JG, Ovod V, Mawuenyega KG, Weiner MW, et al. Validation of Plasma Amyloid-β 42/40 for Detecting Alzheimer Disease Amyloid Plaques. Neurology. 2021 Dec 14;10.1212/WNL.0000000000013211.
28. Udeh-Momoh C, Zheng B, Sandebring-Matton A, Novak G, Kivipelto M, Jönsson L, et al. Blood Derived Amyloid Biomarkers for Alzheimer’s Disease Prevention. J Prev Alzheimers Dis. 2022;9(1):12–21.
29. Barthélemy NR, Li Y, Joseph-Mathurin N, Gordon BA, Hassenstab J, Benzinger TLS, et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat Med. 2020 Mar;26(3):398–407.