
J. Shuren1, P.M. Doraiswamy2
1. Director, Center for Devices and Radiological Health (CDRH), US Food and Drug Administration; 2. Professor of Psychiatry and Geriatrics, Duke University School of Medicine
Corresponding Author: P. Murali Doraiswamy, MBBS, FRCP, Professor of Psychiatry and Geriatrics, Duke University School of Medicine, murali.doraiswamy@duke.edu
J Prev Alz Dis 2022;
Published online March 11, 2022, http://dx.doi.org/10.14283/jpad.2022.28
Digital health technologies, such as smart phone apps and artificial intelligence (AI)-driven platforms, offer promise to provide and enhance clinical treatment and self-care but can also come with risks. The U.S. Food and Drug Administration (FDA) distinguishes three types of software related to medical devices: (1) software that on its own is a device, called Software as a Medical Device (SaMD), (2) software that is integral to a device, called Software in a Medical Device (SiMD), and (3) software used in the manufacture or maintenance of a device. A medical device software program that is deployed on a smart phone or other mobile platform is called a “mobile medical app” (MMA). FDA applies a risk-based approach to its oversight of all devices. FDA generally reviews scientific data to determine whether or not to authorize moderate and high risk devices, including SaMD, for marketing.
While the term ‘digital therapeutics” is sometimes used in the literature to distinguish evidence-based digital health applications from wellness applications, this term is not used for regulatory purposes. Smart phone apps that don’t meet the legal definition of a device do not fall under FDA regulation. For example, a smartphone memory game meant purely for entertainment is not a medical device but a game promoted to improve memory in individuals with Alzheimer’s Disease (AD) would be regulated as a MMA. In the field of neuropsychiatry, software based therapeutics have been cleared by the FDA for treating substance abuse, attention deficit hyperactivity disorder and insomnia (Table 1). To date, no SaMD has been authorized for treating AD or mild cognitive impairment (MCI).
The Center of Devices and Radiological Health (CDRH) is the branch of the FDA responsible for the pre-market approval of medical devices as well as overseeing manufacturing and post-market safety. In this Perspective, Dr. Jeffrey Shuren, the director of CDRH discusses the FDA’s current thinking about digital therapeutics for mild cognitive impairment (MCI) and Alzheimer’s disease (AD) as well as the role of the FDA’s Digital Health Center of Excellence. Dr. Shuren is a behavioral neurologist who has held a variety of leadership roles at the FDA and CMS. This article is adapted from a conversation between Dr. Murali Doraiswamy (MD) and Dr. Jeffrey Shuren (JS) at the 2021 CTAD conference in Boston.

Table 1. Examples of SaMD and SiMD Applications in Neurology / Psychiatry
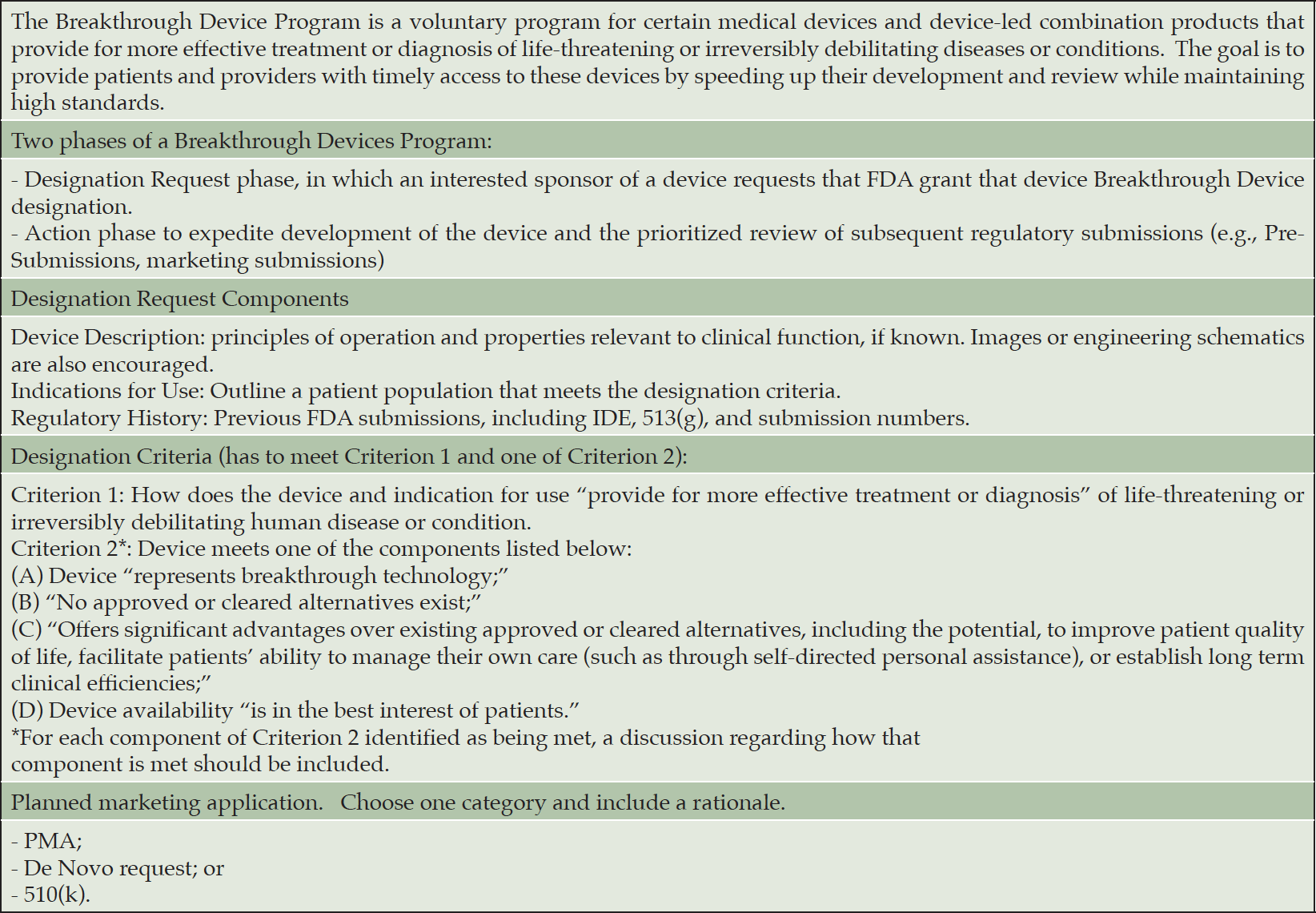
Table 2. Process and Illustrative Example of a Breakthrough Device Designation Request
Source: https://www.fda.gov/media/108135/download
MD: The FDA CDRH plays a key role during pandemics. Can you give us a sense of the scale?
JS: We’ve authorized, through emergency use or full authorization, over 2100 medical devices for COVID-19, including over 400 tests, which is just a remarkable amount of effort. I’m proud of my team and others throughout the FDA.
MD: The pandemic has brought remote care to the forefront. What role do you think smart phones and software devices will play in the future – treatment or prevention— for Alzheimer’s disease (AD)?
JS: Alzheimer’s, like other neurodegenerative diseases, is going to continue to evolve in an individual – their clinical status, their home-life status – it’s just going to change on an ongoing basis. One of the great potential values of digital therapeutics and digital diagnostics is the opportunity to provide interventions in real or near-real time as needed, and to empower caregivers, and, depending upon the state of the disease, sometimes patients themselves, to be able to take actions to better handle their care and their activities of daily living (ADLs); that they can make more targeted decisions and be able to monitor how they are doing in the course of their illness.
MD: Are you optimistic that we’ll have some digital therapeutics soon for Alzheimer’s?
JS: I am! There is certainly a lot of work that’s being done. In the device world right now, unfortunately, there is no technology yet that can slow down the rate of progression of the disease. We’re not there yet, unlike for some other neurodegenerative diseases, like Parkinson’s. But we are hoping we will have those kinds of technologies and if not, clearly things that maybe are helping people in handling their day-to-day activities.
MD: FDA has an “Accelerated Approval” and a “Breakthrough Device” process. Could you explain the rationale, benefits, and possible risks of these processes?
JS: For devices, we have a breakthrough devices program. Accelerated approval is a drug program but the key features of accelerated approval have been incorporated into the breakthrough devices program. We’ve tried to channel a lot of our approaches into one pathway that provides lots of options. The idea behind it is that these are some of the most important technologies for healthcare and as a result, maybe should be treated a little differently. To qualify, or be designated as a breakthrough device, the technology must be intended to treat or diagnose a life-threatening or irreversibly debilitating condition, and, essentially, provide an added benefit to patients. So, you may be addressing a medical need for which there is nothing else on the marketplace, or you may be superior to something else that’s being used as a part of standard of care. And if so, what we offer is the opportunity for far greater engagement with our experts. In fact, we offer the opportunity to engage in what we call “regulatory sprints.” The developer may identify an issue and we will commit with the developer to try to address it within 45 days, and, if not, do another sprint. We learned from COVID-19, too, that one of the game changers for bringing all that great technology to the marketplace was a program that we offered to developers to work with our experts in real or near-real time to have their questions answered, proactively problem solve, and even look at data on a rolling basis. In our breakthrough devices program, we haven’t quite gotten to the COVID-19 level of engagement yet due to insufficient resources, but there’s more interaction than in our other pre-market programs.
MD: How successful has the breakthrough devices program been?
JS: The program is so popular now, and I think the pipeline for great technology in the U.S. is getting better, that the number of devices designated “breakthrough” annually, has increased every single year since we started the program back in 2015. And 2021 beat out 2020, so we now have over 600 devices designated as “breakthrough.” Of those, we’ve already authorized 39 of them with more coming soon. Now, the downside I’ll tell you, is we’ve never been resourced for this work. And as you can imagine, without the resources, our opportunity to work with those developers is a bit at risk. We get funding in part from industry through user fees – not to pay us for decisions, but for enhanced performance. And one of the topics on the table for the upcoming reauthorization of the program is the opportunity for these kinds of breakthrough devices to get the kind of engagement we offered during COVID. It’s not what you get today but more like what you got during COVID but on steroids, which we think could be a true gamechanger in getting those technologies that are safe and effective to market more quickly, and the ones that are not, let them fail faster, so developers can move on.
MD: Several digital therapeutics/apps are being studied by academic consortia, through NIH or foundation grants, to evaluate their benefit in managing Alzheimer’s or MCI. How can the FDA or companies use the results of such academic studies to either create or extend medical claims in this type of regulated environment?
JS: We look at the totality of the evidence. If a developer has access to a study conducted by others on their technology, they are welcome to leverage that in support of a pre-market authorization decision by the FDA. We will also look at published literature. The challenge becomes that sometimes, to really understand what is going on with that device, we need to drill down into the data itself. Therefore, sometimes, we can’t do a lot with a published study unless we get the raw data. If we can, we’ll leverage that to support our decision making. We’ve done that in several instances.
MD: How do you view what is a “clinically meaningful” benefit for MCI or Alzheimer’s in a device/app trial?
JS: That’s a great question, and in part, because we don’t have the technology today to slow down the progression of the disease, although there are folks working on digital therapeutics using some unique approaches, such as using sound and light, so we’ll see what happens. But probably the most important measure today for what we are dealing with is if you have a significant impact on ADLs – can you keep someone independent longer? Are you able to facilitate communication? What about an impact on how people feel? Of course, clinically meaningful benefit can range widely, and we will always look at the benefit in the context of the risks posed by the device. So we encourage developers, in whatever you’re doing, to please take advantage of our pre-submission meeting program, which offers an opportunity to present to us what you’re thinking or what you think the technology is going to do and questions or issues you want our advice on, and we can work together on figuring out the right metrics to show effectiveness and to look at safety.
MD: Is there an opportunity for device/app developers to distinguish between say, short term symptomatic benefits versus long-term disease modifying benefits?
JS: There is, and again, we look to “What is the technology intended to do?” But we’ll also go further. You know, sometimes the developer gathers their data, and it may not support the use that they thought the technology could be used for. However, the data may support a different use or modified use of the device in which case, fine, we’ll grant an authorization for that different use, and if new data arises in the future to support other indications, we’ll go ahead and authorize it for those uses, as well.
MD: Is age-associated cognitive decline within the CDRH’s purview? Can someone seek a claim for an app/device to improve age-related memory problems?
JS: Obviously, there’s that line between wellness and disease actions/uses, which we have guidance on, that we don’t regulate. When we start getting into clinical states, disease states, those are indications we regulate. My recommendation for any developer looking at that boundary, come talk to us. It’s about what you want to do is to and we can work with you as to what are the right steps to take. If it turns out it’s something that is not under our purview, we’ll tell you.
MD: With many digital health devices/apps the same technology may help healthy populations as well as clinical populations. But they can only be marketed for one or the other. Example maybe an app for improving normal sleep versus clinical insomnia. How does the CDRH view such dual use technologies? How should scientists or companies with such dual use technologies approach the FDA?
JS: Typically, dual use, as you note, is where a technology is for indications that are a “non-medical device” claim, which could be a wellness claim, and a medical device claim. We have policy in a guidance that talks about this dual use. When the technology, or part of the technology, let’s say, is for the non-medical device indication, we will not look at that. We won’t be asking for data to support the non-medical device indication, unless the technology or a modification to that technology, could impact its performance for the medical device indication. In that circumstance, we will want to look under the hood of that part of the technology, but only as it relates to the device indication.
MD: Your office highlighted some 90 examples of how real-world data has been used in the regulatory process. How can real world data, generated outside of a randomized trial, be helpful for regulatory submissions in the Alzheimer’s/MCI field?
JS: We put out those examples to show folks the wide variety of the kinds of decisions for which that kind of evidence can be used to support. So, the 90 examples cover not only post-market decisions but also pre-market decisions. And we have seen over time that the number of instances where we can leverage such evidence continues to increase from year to year, and it covers a wide range of products. What matters for our ability to leverage such evidence is two factors – and we put this out in a guidance back in 2017, I think – which are relevance and reliability. Is the data relevant for the decision we are making, for the question we are trying to answer? And is it sufficiently reliable? We lay out a variety of factors that could be considered in making that kind of decision. Certainly, real world data can be part of a traditional clinical trial, if you will, leveraging from real world data sources. But we leverage such data outside of those circumstances, too. If you think about it, a traditional clinical trial, such as a randomized, controlled trial –employs a scientific methodology, as you well know, to reduce bias, so we have higher confidence in the results. The challenge of course is, the more bells and whistles you put into a study, and the more types of patients you exclude, the less that population who’s evaluated may represent the people who are in fact going to use that technology, or have it used on them once it’s out there in the wild, in clinical use. As a result, so often with devices, you don’t understand the true benefit-risk profile of the technology until it’s being used in routine clinical care. Having real world data sources in which you have high confidence in the quality of the data provides an opportunity to gather that kind of information. As you mention, large pragmatic trials are a way to address these issues of bias, and reflect the populations who are going to use that device and give you a more realistic understanding of the true benefit-risk profile of the technology and better insight on how to use that technology safely.
MD: Are minority/underserved populations being represented appropriately in device trials?
JS: That’s an important question. It’s been a mix. In some cases, we’ve seen that, and in other cases, no, the trials are not reflective of the broader populations who use or may use that device. In some cases, that may be less important than others. But clearly, there are technologies where there may be differences in performance when used in different populations. More recently, you saw that play out with pulse oximeters, where people who have more skin pigmentation may see a decrement in performance, and it certainly has raised the question that if you’re going to put these products on the marketplace, there should be an expectation that you are assessing a sufficient number of people who have darker pigmented skin, for that reason, and those are steps that we have taken at the center. And yes, there is a greater focus today than in the past, in assuring that technologies are assessed in the appropriate populations. In places where that isn’t yet the case, we want to, at a minimum, move towards transparency about the populations in which the device was tested and not tested, and what we know about performance. You could see this as more of a bridge, an interim step. Ultimately, though, the answer is to get the data.
MD: How does one best assess technology in a range of populations?
JS: One of the challenges we face with technology is the time and cost it can take to assess technology in a large range of different populations. We know that that can have an impact on whether we’ll see the technology hit the marketplace at all. So, part of our work is trying to reduce the time and cost of evidence generation. You had mentioned “real world data” and “real world evidence.” Part of our interest in them is, if there are data collection mechanisms that already exist for which data will be collected, let’s say as part of routine clinical care, and if we can leverage them, we can potentially reduce the time and cost to gather evidence on technology. And why this matters is that we have to think about that “sweet spot” of being able to incentivize innovation to get the new technologies we need, but to also drive the evidence generation to not only support it coming to the market, but for us to have a full understanding of its performance in different populations. That needle between the expectations of what evidence it takes to come to market versus what it may take to stay on the market may, in fact, move a little bit depending upon the importance of the technology and the feasibility of gathering post-market data quickly because the last thing we want to do is put a device out there without knowing it works in the populations who will use it. Nor do we want to have to say we only have data on its use in one population and then have the device out there being used off-label in other populations. You want to get the evidence. By the same token, we’ve got to figure out how to generate the evidence in ways that don’t stifle the development of the technology, in the first place, such as due to untenably high costs. This is of big importance to us, to spur innovation, but make sure that we get technology that’s available for all people.
MD: The CDRH has a flagship Digital Health Center of Excellence. Can you tell us about it? And can you share your advice for how the Alzheimer’s field can help spur digital innovation?
JS: I’m very excited about the Center of Excellence, which we launched last fall. It is an opportunity to truly advance the work that we do in digital health, by bringing together in a virtual center our core experts that sit in our division of digital health with experts around CDRH who are participating in digital health – including in cybersecurity and related fields – all into one virtual entity for the purpose of better leveraging our expertise to drive greater innovation, to be more consistent, and predictable and efficient in our work, to serve as a resource within our center as well as to the rest of the FDA and to our external stakeholders. In addition, to drive and do a better job at driving a lot of the science, we need to help facilitate bringing technologies to market and to serve as a test bed for innovative regulatory approaches for digital health technologies. The framework Congress put in place for devices, including digital health technologies, is over four decades old. I mean it literally was designed for my grandmother’s technology! Most of it was hardware-based, but today we have many software-based devices, which, as you noted, is called in international terminology, software as a medical device or SaMD. And of course, those technologies are just, different. They have more rapid innovation cycles. In fact, if you hold up making certain changes to those technologies, you put people at risk. You need to make a lot of changes to keep that device safe, such as to address cybersecurity vulnerabilities. So, we need to think about new regulatory paradigms that are better tailored to SaMD. This center of excellence has been test driving some of them. One such approach, that we call “precertification,” is focused on accessing the capabilities of the developer and the extent to which we can leverage that understanding in our reviews, as opposed to just focusing on the technology, in order to expedite products coming to the marketplace, and then build in a post-market feedback loop that leverages real-world data collection, maybe even leveraging data collected by the technology itself. Today devices are playing an increasingly important part in data collection as a part of clinical evidence generation. This Center of Excellence is serving a critical role in that capacity. In fact, there are legislative proposals that, if Congress ultimately supports them, we could have a modern regulatory framework to support digital health technologies. And the Center of Excellence will be leading the charge.
MD: How can we spur digital health innovation?
JS: In terms of spurring innovation, one approach is to have a regulatory paradigm that’s better suited for these devices. Because if you set the bars in the right place, and we’re not talking about changing the US authorization standard of safety and effectiveness, but instead providing greater flexibility on how that standard is met and better tailored to the technology, this creates efficiencies, reduces unnecessary costs, and makes it more attractive to have innovative technology come to the U.S. And I’d love to see better reimbursement available, as well, because we know that will drive great technologies coming to the marketplace. And, of course, we’ll see what happens with our user fee negotiations if we get the funding to support this pilot to have greater interaction with developers of breakthrough devices, something we call “the total product life cycle advisory program,” or TAP for short. If we get TAP, we think we can advance these technologies and if Congress enacts a more modern regulatory framework, then, from an FDA standpoint, I’d say we are in fantastic shape. And if we can get payment policies to come into alignment, I think the U.S. would be incredibly attractive for innovative technology to be developed and available, and that includes in the Alzheimer’s space.
MD: That’s inspiring. You’ve taught us a lot. Thank you for your time.
Disclosures/Acknowledgment: JS has no conflicts of interest to report. PMD has received grants, advisory fees, and/or stock from biotechnology companies and is a coinventor on patents for the diagnosis and treatment of dementia.