
L.S. Schneider1, M. Laudon2, T. Nir2, J. Caceres3, G. Ianniciello3, M. Capulli3, N. Zisapel2,4
1. Department of Psychiatry and the Behavioral Sciences, and Department of Neurology, Keck School of Medicine, and Leonard Davis School of Gerontology of the University of Southern California, Los Angeles, CA, USA; 2. Neurim Pharmaceuticals Ltd, Tel Aviv, Israel; 3. Dante Labs, Cambridge , United Kingdom; 4. Department of Neurobiology, Faculty of Life Sciences, Tel-Aviv University, Tel Aviv, Israel
Corresponding Author: Lon S. Schneider, Department of Psychiatry and the Behavioral Sciences, and Department of Neurology, Keck School of Medicine, and Leonard Davis School of Gerontology of the University of Southern California, Los Angeles, CA, USA, lschneid@usc.edu
J Prev Alz Dis 2021;
Published online November 23, 2021, http://dx.doi.org/10.14283/jpad.2021.61
Abstract
Background: Piromelatine is a novel melatonin MT1/2/3 and serotonin 5-HT-1A/1D receptors agonist developed for mild Alzheimer’s disease (AD). In a randomized, placebo-controlled, dose-ranging study (ReCognition) of piromelatine (5, 20, and 50 mg daily for 6 months) in participants with mild dementia due to AD (n=371, age 60-85 years), no statistically significant differences were found between the piromelatine and placebo-treated groups on the primary (i.e., computerized neuropsychological test battery (cNTB)) and secondary outcomes (ADCS-CGIC, ADCS-MCI-ADL, ADAS-cog14, NPI, and Pittsburgh Sleep Quality Index (PSQI)) nor were there safety concerns (https://clinicaltrials.gov/ct2/show/NCT02615002).
Objectives: This study was aimed at identifying genetic markers predicting piromelatine treatment response using a genome-wide association approach (GWAS).
Design: Variant genotyping of a combined whole genome and whole exome sequencing was performed using DNA extracted from lymphocytes from consenting participants. The general case-control allelic test was performed on piromelatine-treated participants, taking “responders” (i.e., >0.125 change from baseline in the cNTB) as cases and “non responders” as controls, using a Cochran-Armitage trend test.
Setting: 58 outpatient clinics in the US.
Participants: 371 participants were randomized in the trial; 107 provided informed consent for genotyping.
Results: The GWAS sample did not differ from the full study cohort in demographics, baseline characteristics, or response to piromelatine. Six single-nucleotide polymorphisms (SNPs) in chromosome 2q12 (2:107,510,000-107,540,000) were associated with response (p-value < 1×10 -4 each). Post hoc analyses suggested that the carriers of the 2q12 polymorphism cluster (27% of the GWAS sample) improved significantly on the cNTB on piromelatine as compared to placebo but significantly worsened on the ADAS-Cog14 and PSQI. By contrast, “non-carriers” improved significantly with piromelatine compared to placebo on the ADAS-Cog14 ( 2.91 (N=23) with piromelatine 20 mg vs 1.65 (N=19) with placebo (p=0.03)) and PSQI.
Conclusions: The 2q12 (2:107,510,000-107,540,000) 5-6 SNPs cluster may predict efficacy of piromelatine for mild AD. These findings warrant further investigation in a larger, prospective early-stage AD clinical trial for patients who are non-carriers of the 2q12 polymorphism cluster.
Key words: Piromelatine, Alzheimer’s disease, GWAS, SNP, clinical trials, ADAS-cog, sleep, cognition.
Introduction
Current marketed drugs for Alzheimer’s disease (AD) include cholinesterase inhibitors and memantine and are modestly effective over a limited time. Despite enormous efforts of pharmaceutical companies and academic organizations, means of preventing, delaying the onset, slowing the progression, and improving the symptoms of AD are urgently needed. There have been repeated failures in developing effectives treatments aimed at various targets, including anti- amyloid β-protein (Aβ) and anti-tau interventions, anti-neuroinflammation and neuroprotection interventions. The recent approval by FDA of the anti-amyloid-beta antibody, aducanumab, was based on its effect on reducing amyloid beta plaque, and the requirement for a post-marketing clinical trial to eventually determine clinical benefit (1). (https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2021/761178Orig1s000ltr.pdf).
AD patients exhibit severe sleep/wake disturbances and insomnia, and these are associated with more rapid cognitive decline and memory impairment (2). Early neuropathological changes in AD are accompanied by decreased CSF melatonin levels (3). The reduced melatonin levels are already found in the preclinical stages and correlate significantly with the severity of mental and sleep impairments in patients with dementia (4). Piromelatine (N-(2-(5-methoxy-1H-indol-3-yl) ethyl)-4-oxo-4H-pyran-2-carboxamide), is a novel melatonin MT1/2/3 and serotonin 5-HT-1A/1D receptors agonist developed for mild AD. Melatonin receptor agonism appears to clinically benefit mild to moderate AD patients (4). Simultaneous activation of melatoninergic and 5-HT-1A receptors may synergistically promote neurogenesis (5). Piromelatine, 20 mg and 50 mg daily over 1 month enhanced sleep maintenance in a phase 2, randomized, placebo-controlled, sleep study in patients with insomnia (n=137 aged 18-80) and enhanced NREM delta power and decreased beta power (6). Both effects may be beneficial in early AD and enhance Aβ clearance from the brain (7, 8).
In a phase 2, randomized, placebo-controlled, dose-ranging trial (ReCognition, NCT02615002 (9)) of piromelatine (5, 20, and 50 mg daily for 6 months) in participants with mild dementia due to AD (n=371 age 60-85 years), no statistically significant differences were found between the piromelatine and placebo groups on the primary outcome (computerized Neuropsychological Test Battery (cNTB) (10)) and secondary outcomes, including the Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-cog14) (11) and Pittsburgh Sleep Quality Index (PSQI) (12)), nor were there safety concerns. However, it was noted that 42% of subjects in the 20 mg piromelatine treated groups were categorized as “responders” based on a > 0.125 change from baseline on the cNTB.
Usual medical practice applies the same treatments for all individuals with the targeted disease despite differences between patient’s genetic make-up and the influence of environmental factors. However, it is unreasonable to expect conditions with clinical and pathophysiological diversity to have a simply definable pathological mechanism. In fields such as oncology and hematology molecular testing is routinely used in clinical settings to select more effective treatments and to reduce adverse effects (13).
Genetic diversity contributes to both disease susceptibility and variability in drug response. With the advent of genome-wide association studies (GWAS), screening for individuals with small variations within the genes (single nucleotide polymorphisms; SNPs) can be used for predicting treatment response, allowing more efficient development of new medications, and result in favorable responses to treatment[14]. This study was aimed at identifying genetic factors predicting piromelatine treatment outcome in participants from the phase 2 piromelatine trial (Recognition study) using a GWAS approach. We report on the overall outcomes of the trial (both the total population and GWAS sample), the identification of genetic markers of response and the subpopulation of the GWAS sample who were carriers and non-carriers of the identified genetic marker.
Methods
ReCognition trial design and participants
The protocol, informed consents and amendments were approved by the local institutional review boards (IRB) at 58 sites in the US. Potentially eligible participants or their legally authorized representatives provided written informed consent before participating. Participants or their LARs were also asked to provide informed consent for apolipoprotein-E (ApoE4) isotyping and, further, to provide consent for whole genome sequencing by separate applicable informed consent form. The latter consents were not requirements for trial participation. The trial was registered under ClinicalTrials.gov Identifier: NCT02615002.
A total of 371 male and female outpatients (ages 60-85) diagnosed with mild dementia due to AD for at least 6 months, with Mini-Mental State Examination (MMSE) score of 20 to 27 and Clinical Dementia Rating Global Score (CDR) of 0.5 or 1 (mild dementia) were randomized and 304 patients completed the clinical trial. Participants had to have no evidence of focal disease to account for dementia as established by computed tomography (CT) or magnetic resonance imaging (MRI) scans. They had to be on stable doses of an acetylcholinesterase inhibitor (with or without memantine) for 4 months prior to baseline. Those using melatonin, benzodiazepines, or other hypnotics during the 2 weeks before baseline or during run-in were excluded. Following diagnosis, all patients underwent a 2-week single blind placebo run-in, followed by double-blind randomization to treatment with piromelatine (5, 20, and 50 mg) or matching placebo tablets for 26 weeks. A telephone follow-up was completed 2 weeks after the last dose of study medication to elicit any safety concerns. The treatment was double-blinded, with four parallel treatment groups. Randomization was assigned after successful completion of the single-blind run-in period using an interactive web response system (IWRS), with an allocation ratio of 1.2 (placebo):1:1:1 to each treatment group. Study medication was administered orally, one tablet/day, 1-2 hours before bedtime, preferably between 21:00 h and 23:00 h, after the dinner. In addition, participants were instructed to spend 2 hours a day in outdoor daylight.
The PSQI was completed by the study staff with input from both the study partner and patient. Conflicting information was resolved in favor of the study partner’s responses. Safety parameters were assessed at each visit and included spontaneously reported Adverse Events (AEs) or serious AEs (SAEs) and vital signs (heart rate and blood pressure), physical examination, laboratory tests (hematology, biochemistry [including prolactin and testosterone] and urinalysis) and 12-lead ECG.
Based on an estimated effect size for the cNTB between treatment dose and placebo of 0.35 over 26 weeks, a significance level, α = 0.05 for one-sided test, and power (1-β) = 88%, a sample size of 115 patients for the placebo arm and 95 patients for each of the 3 piromelatine arms was calculated. Assuming a 50% screen failure rate and allowing for 15% patient withdrawal, 920 patients were to be screened to randomly assign 460 patients, of whom it was expected that 400 would complete the study.
The primary objective was to compare the effect of piromelatine (5, 20, and 50 mg) to that of placebo on the global composite score of the cNTB (10) after 26 weeks of double-blind treatment. The global composite score includes the International Shopping List Test (ISLT; immediate and delayed recall), One Card Learning (OCL), Identification (IDN), Detection (DET), and One Back Card (OBK), Controlled Oral Word Association Test (COWAT), and Category Fluency Test (CFT). Secondary objectives were to compare the effect of piromelatine (5, 20, and 50 mg) to that of placebo on global impression assessed by the Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change (ADCS-CGIC)(15), the ADCS-Activities of Daily Living scale, mild cognitive impairment version (ADCS-MCI-ADL) (16) and the ADAS-cog14 (11) also after 26 weeks. Sleep quality, an exploratory endpoint, was assessed via the PSQI (12).
Statistical analysis: Following database lock results were summarized and analyzed according to the pre-planned statistical analysis plan. Primary and secondary efficacy analyses were carried out using an analysis of covariance (ANCOVA) model with treatment and study site as the main effects and baseline cNTB global composite scores, age, gender, and ApoE4 as covariates. Missing data (at the level of the composite score) were imputed using a pattern mixture model approach.
For the safety analysis set (i.e., all patients who were randomized to treatment and who took at least one dose of the study medication) and full analysis set (FAS; all patients in the safety analysis set who satisfied all entry criteria and who had efficacy data for the primary outcome at baseline and at least one post baseline assessment). Differences between groups were tested at 5% one-sided significance level to achieve 80% power. The data was analyzed using the SAS ® version 9.4 (SAS® Institute Inc., Cary, North Carolina, USA).
Genome wide association study (GWAS)
For participants in the GWAS, whole blood was collected at the time of enrollment, and genomic DNA was extracted from peripheral lymphocytes using a commercial kit to be used for APOE isotyping. For the 107 patients in the GWAS sample variant genotyping of a combined Whole Genome and Whole Exome sequencing was performed. The detection of variants associated with the response of the treatment was performed through a GWAS analysis (case-control paradigm) on the piromelatine treated groups with the highest numbers of cNTB responders. The setup involved taking the patients that respond to the treatment (i.e., > 0.125 change from baseline in cNTB) as case and the patients that did not respond to the treatment as controls. The general case-control allelic test was performed through a Cochran-Armitage trend test. These results were cross-checked against potential confounding variables, given as a stratification test. The stratification was assessed by a complete-linkage hierarchical clustering using the pairwise population concordance as a constraint. The SNP calling was done on the aligned BAM files using DRAGEN and filter SNPs using VQSR (17).
The SNP caller program takes mapped and aligned DNA reads as input and calls SNPs and indels through a combination of column-wise detection and local de novo assembly of haplotypes. Callable reference regions are first identified with sufficient alignment coverage. Within these reference regions, a fast scan of the sorted reads identifies active regions, centered around pileup columns with evidence of a variant. The active regions are padded with enough context to cover significant, nonreference content nearby and padded even more where there is evidence of indels.
Statistical analysis
For post-hoc analyses of the GWAS sample, baseline and efficacy results between the FAS and the GWAS sample were compared using t-tests. Following identification of candidate genetic markers in the GWAS sample the efficacy data for the “carriers” and “non-carriers” subpopulations were analyzed. Given the exploratory nature of the study, P-values are reported without adjustment for multiplicity.
Results
A total of 371 participants were randomized into the treatment groups: 106 in the placebo group, 89 in the piromelatine 5-mg group, 88 in the piromelatine 20-mg group, and 88 in the piromelatine 50-mg group. A total of 352 were included in the FAS and 304 (81.9%) completed this study (Table 1). The mean age was 73.1 years (SD 7.1, range, 60 to 85 years), 200 (56.8%) female. Most participants (88.6%) were white and not Hispanic or Latino (76.4%). At baseline 224 patients (63.6%) were on cholinesterase inhibitors (with or without memantine) and 128 patients (36.4%) were treatment naïve at baseline.
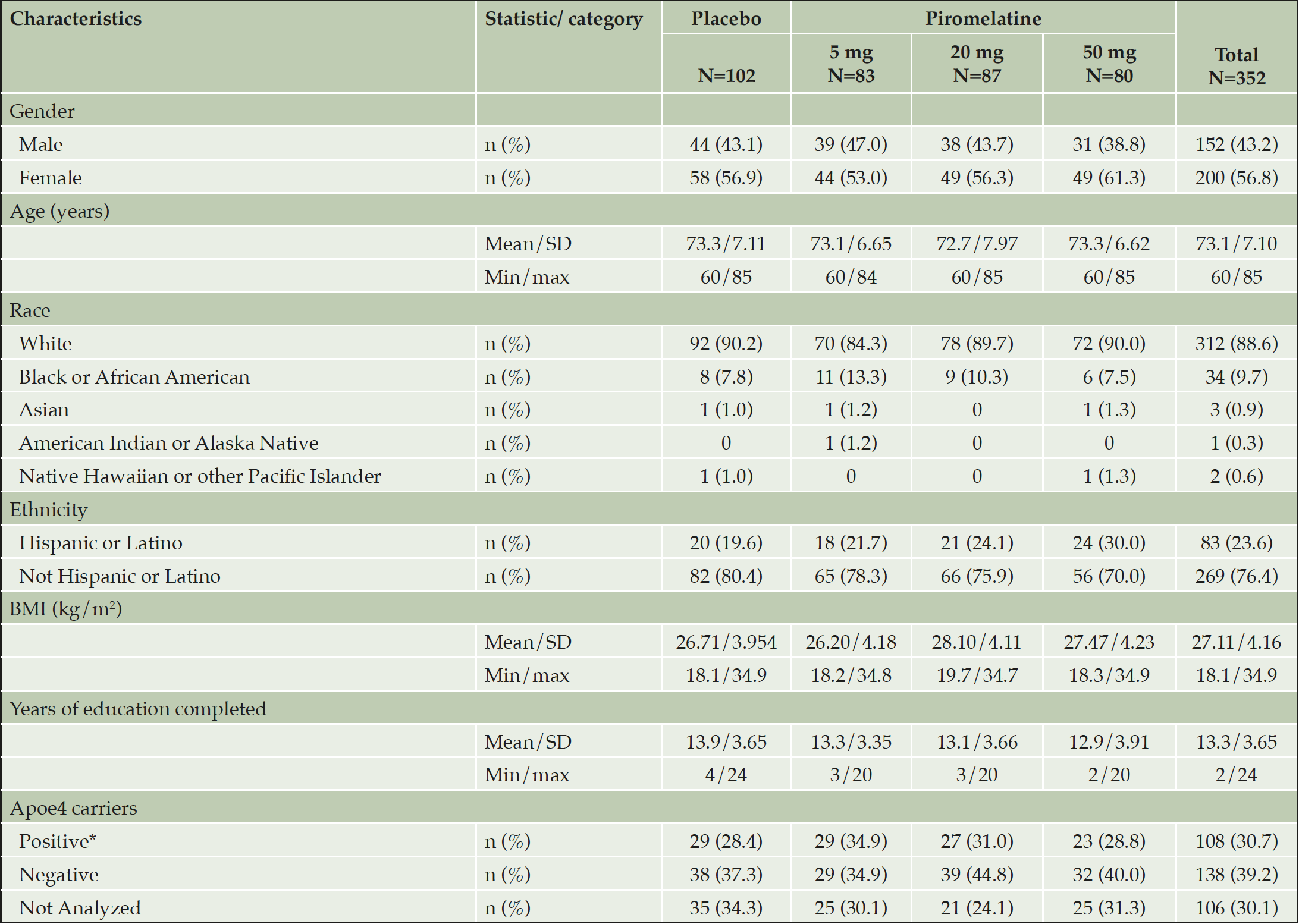
Table 1. Demographics for Patients (Full Analysis Set)
Abbreviations: BMI=body mass index, max=maximum, min=minimum, SD=standard deviation. *ApoE4 carrier status (the presence of 1 or more copy(ies) of the ApoE4 allele).
There were no statistically significant differences between placebo and piromelatine (5mg, 20mg, and 50mg) treatment arms in the cNTB global composite scores (Table 2). Similarly, no significant differences were observed between placebo and piromelatine arms in the secondary outcomes (Table 2). Overall, no safety concerns were observed in this study and no specific trends in adverse events by treatment group or piromelatine dose were noted and no difference between the placebo and piromelatine groups (Table 3).
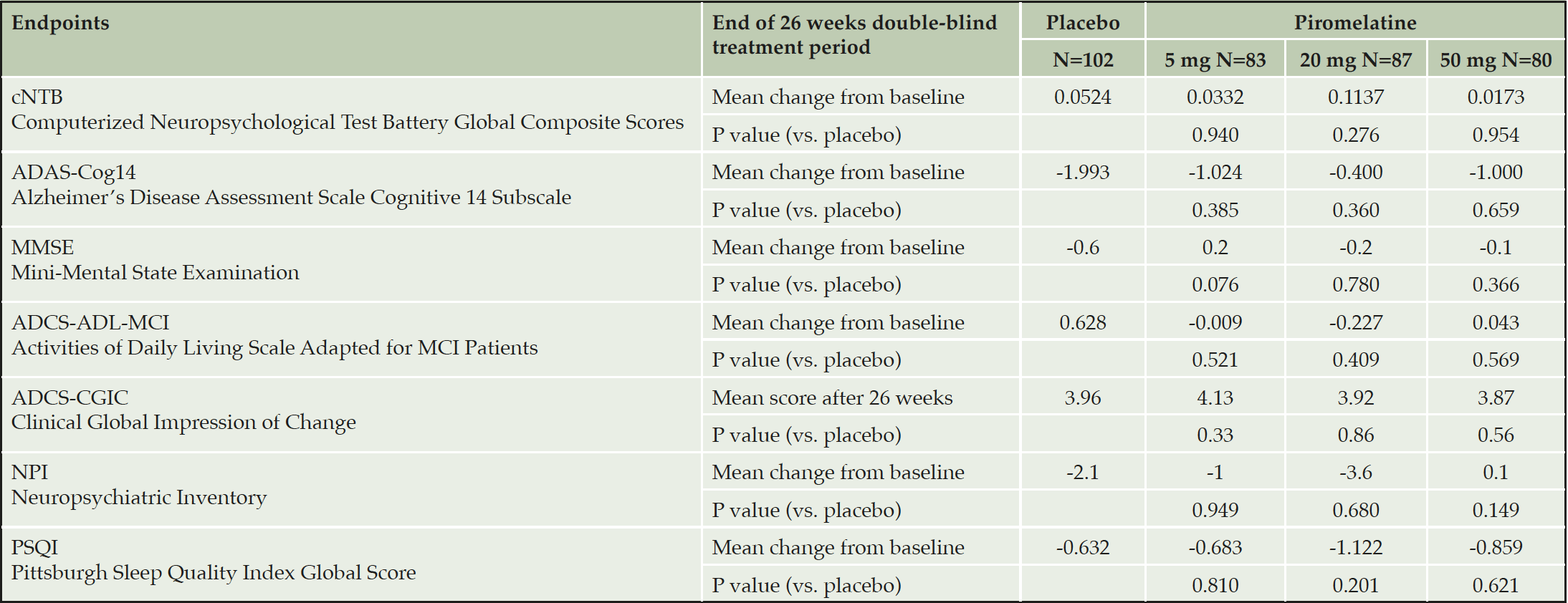
Table 2. Overview of Treatment Differences after 26 weeks of double blind piromelatine and placebo treatment in patients with mild AD (Full Analysis Set)
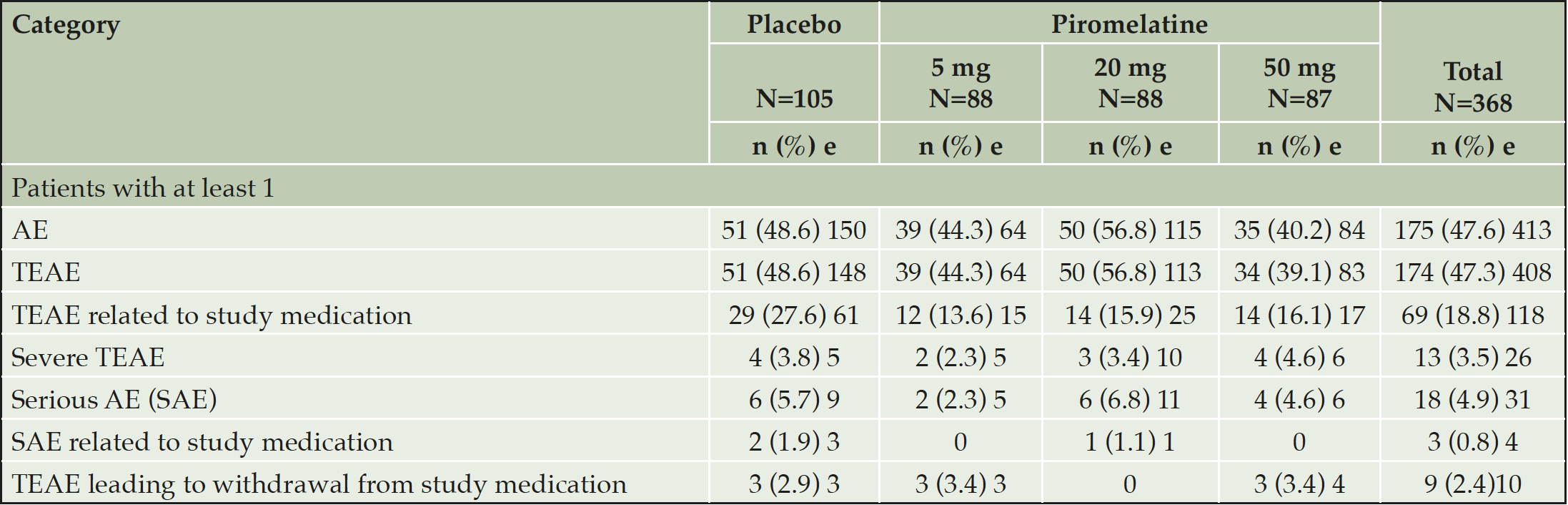
Table 3. All Adverse Events (Safety Analysis Set)
AE=adverse event, e=number of adverse events, N=number of subjects in the group, n=number of subjects in the specified category, TEAE=treatment-emergent adverse event.
The threshold of discrimination for changes in health-related quality of life for chronic diseases has been suggested to be approximately half a standard deviation (SD) (18). Accordingly, the cNTB responder criteria used a threshold of 0.125 (half of the SD of the mean change from baseline in cNTB after 26 weeks of treatment). It was found that the responder rates to piromelatine 5, 20 and 50 mg doses in the FAS were 40, 42 and 37%, respectively.
A total of 107 participants (from 33 of the 58 clinical sites) of 304 who completed the 6 months double-blind treatment period consented to whole genome sequencing and participated in this study. To determine if the GWAS sample was representative of the FAS we compared demographics, baseline characteristics and treatment effects between these cohorts. As can be seen in Table 4, the GWAS and FAS samples did not differ in baseline characteristics or treatment effects. (Similar results were found when the GWAS sample was compared with the FAS minus GWAS sample subpopulation).
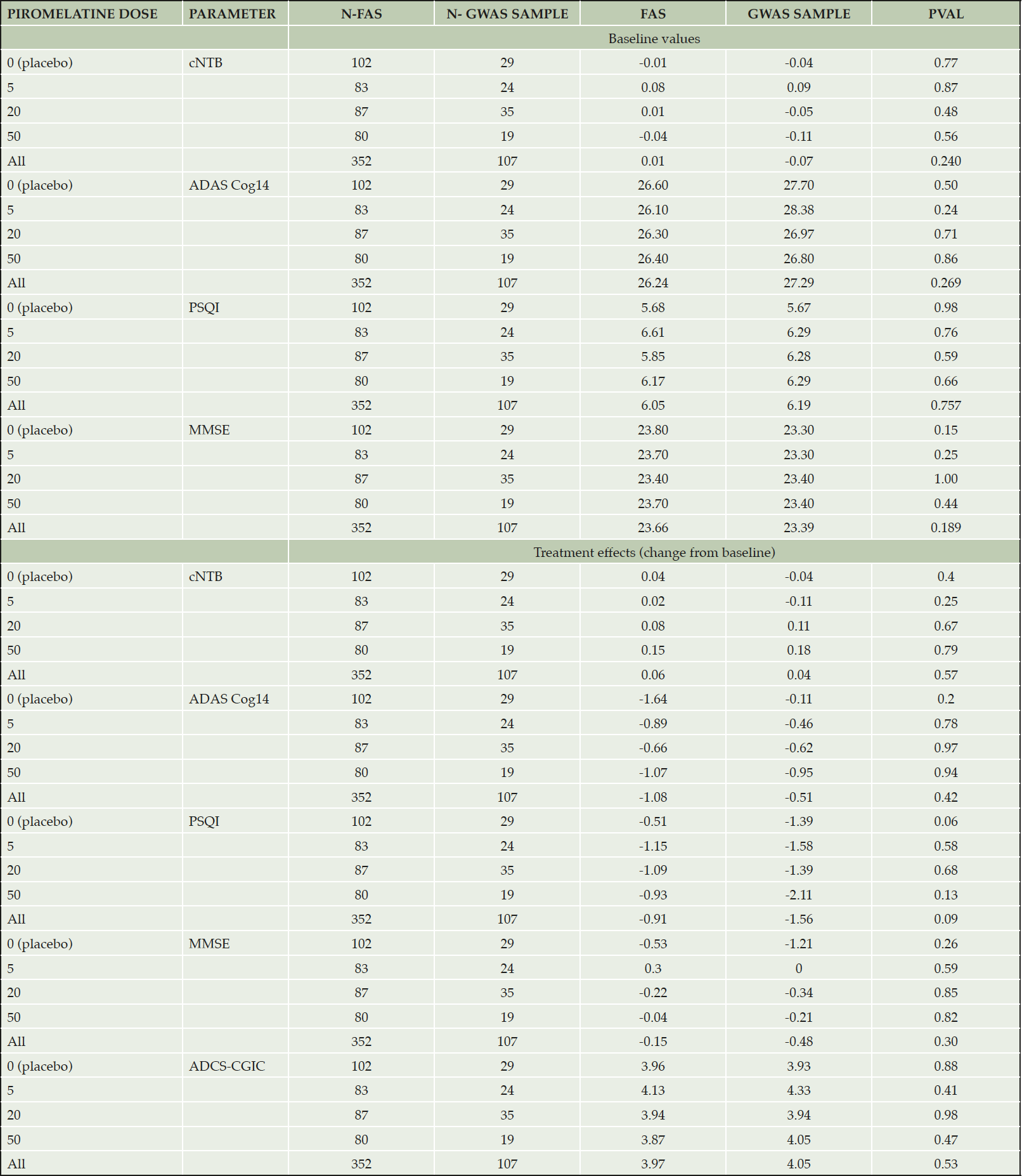
Table 4. Baseline values and treatment effects (piromelatine and placebo) in the FAS and GWAS cohorts
Based on the insignificant differences in baseline values and treatment response, the GWAS sample was considered representative of the ReCognition FAS. GWAS was then performed for cNTB responders vs non-responders in the GWAS sample for subjects treated with the 5 and 20 piromelatine dose (59 participants) that had the largest percentage of responders in the FAS, leaving out the 50 mg and placebo treated groups for further validation of the results. SNPs that did not pass quality control filtering were excluded.
We did not observe any SNP p-value with enough significance in isolation to ensure the discovery of a locus associated with drug response. However, in the primary GWAS cycle we observed a few loci in chromosome 2q12, within the range 2:107,510,000-107,540,000 which had multiple SNPs with a p-value < 1×10-4 each (rs12328439 (T>C), rs62155556 (T>A, rs62155557 (G>T), rs62155558 (G>A), rs17033479 (A>G) and rs9789618 (T>A), as disclosed in the Genome Reference Consortium Human genome build 37 (GRCh37)). In particular, the SNPs in this locus also appear the most significant list as the best predictors of response when adjusted for covariates (apoE alleles, piromelatine dose, and group) which added further significance to the loci.
Subsequent analyses suggested that the participants carrying 5-6 SNPs of the 2q12 polymorphism cluster, 27% of the GWAS sample (there were no participants with less than 5 SNPs in the cohort), did not differ from non-carriers on baseline characteristics but differed significantly in response to piromelatine and placebo (Figure 1, Table 5).
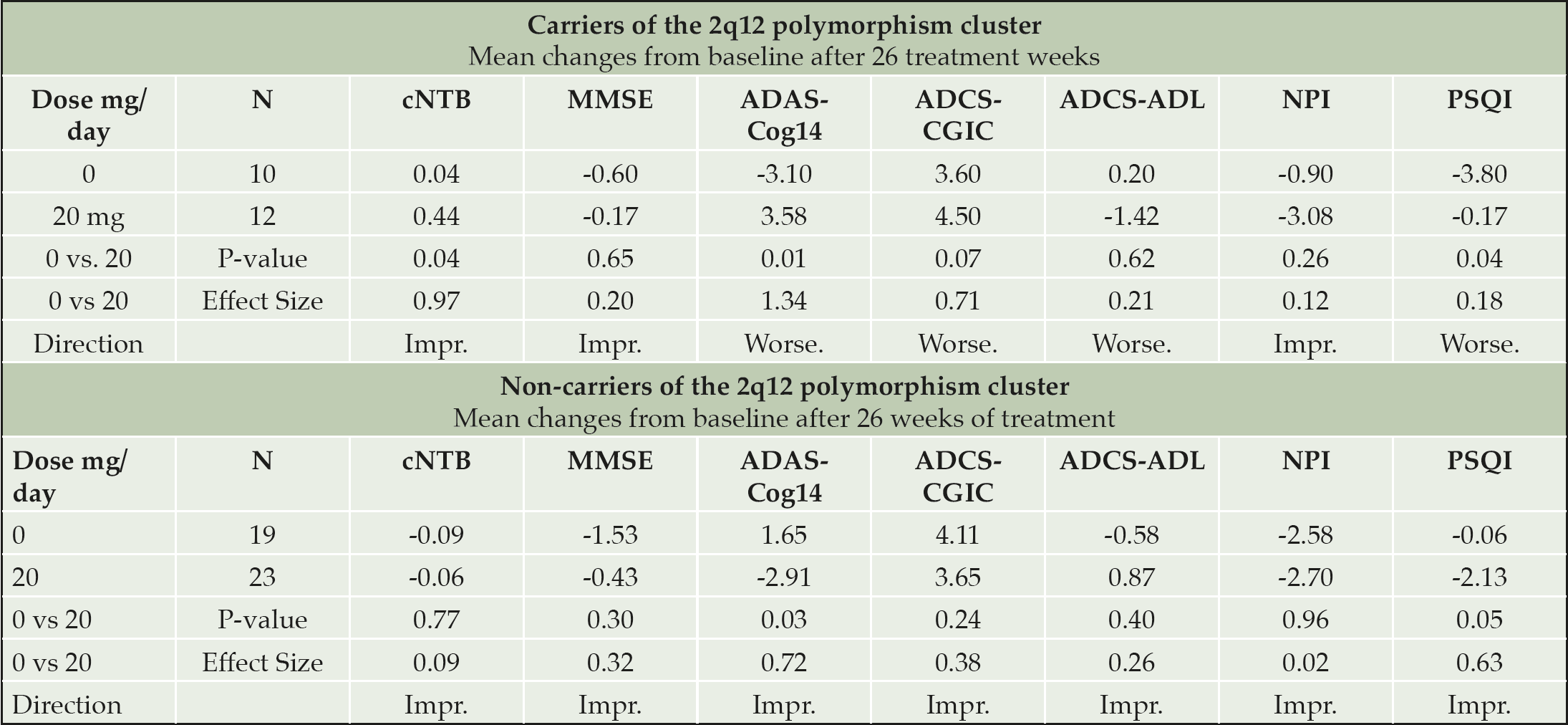
Table 5. Effects of piromelatine 20 mg and placebo (26 weeks) on cognitive parameters and other measures in Carriers and non-carriers of the 2q12 polymorphism cluster
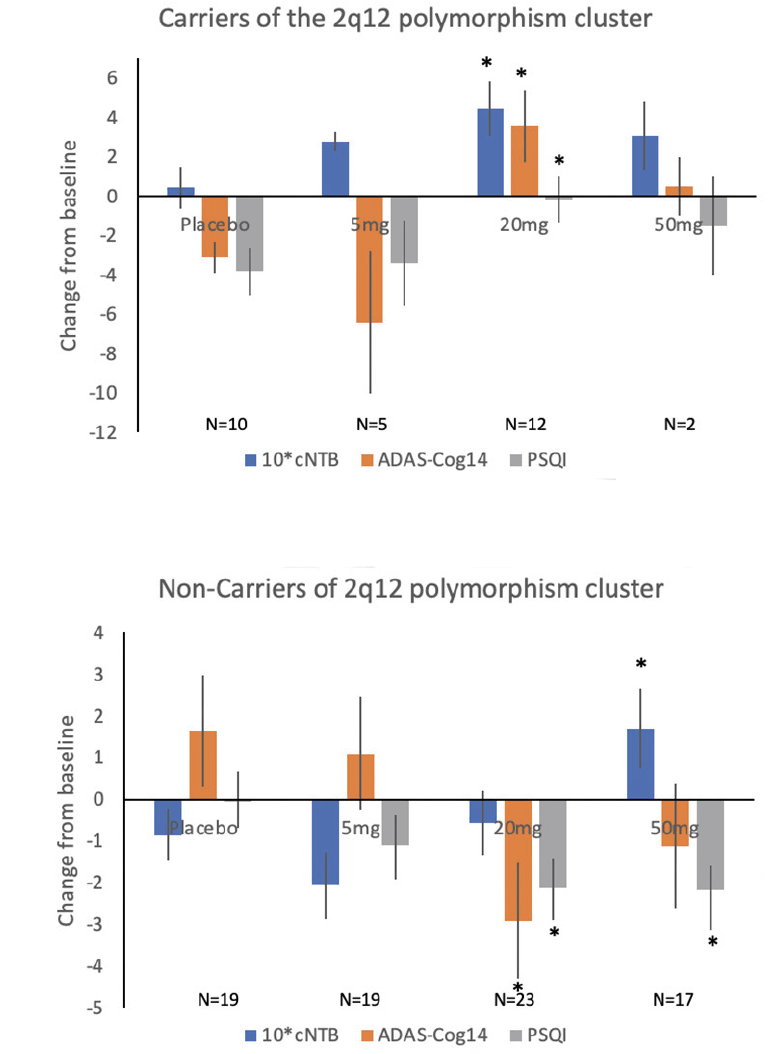
Figure 1. Dose Response (change from baseline +SEM) of Carriers and non-carriers of the 2q12 polymorphism cluster to piromelatine and placebo (26 weeks)
Carriers of the 2q12 polymorphism cluster (27% of the GWAS sample) improved significantly on the cNTB with piromelatine at 26 weeks as compared to placebo; and as can be seen in Figure 1, the 20 mg dose was the most effective (0.44 (N=12) vs 0.04 (N=10) respectively, p=0.04) (Table 5). However, there was a significant worsening in ADAS-Cog14 (+3.58 (N=12) vs -3.1 (N=10), respectively; p=0.01) and in PSQI (-0.17 (N=12) vs -3.80 (N=10), respectively; p=0.04) (Table 5) in the carriers of this polymorphism cluster. The worsening in ADAS-Cog14 following 26 weeks treatment of 20 mg piromelatine was correlated with worsening in PSQI (R=0.57 p<0.05).
By contrast, the non-carriers of the 2q12 polymorphism cluster (73% of the GWAS sample) improved significantly on the ADAS-Cog14 and PSQI after 26 weeks of piromelatine compared to placebo. The 20 mg dose was significantly more effective in improving ADAS-Cog14 (-2.91 (N=23) vs 1.65 (N=19), respectively; p=0.03) and PSQI (-2.13 (N=23) vs -0.06 (N=19), respectively; p=0.05) (Figure 1 and Table 4). Of the “non-carriers’ cohort,” 34.7 % improved (decreased) by ≥ 4 ADAS-Cog14 points in the piromelatine 20 mg vs. only 11.8% in the placebo group (NNT=4.4).
Discussion
Pharmacogenomics and biomarkers have been used for improving the benefit/risk ratios of drugs and treatment across medicine. In Alzheimer’s drug development most phase 2 and 3 clinical trials failed to show clinical benefit. To overcome this circumstance, the importance of using biomarkers for enhancing recruitment and potential treatment efficacy is well-recognized (e.g. APOE-4 carriers may respond differently than APOE-3 carriers to treatments (19, 20)). In this study, we identified several SNPs associated with a differential response to piromelatine treatment in patients with mild AD. The SNPs identified had not been previously reported to be associated with AD or with response to therapeutic agents.
The results suggest that the 5-6 2q12 (2:107,510,000-107,540,000) 5-6 SNPs polymorphism cluster may serve as a predictor of cognitive enhancement with piromelatine treatment measured by cNTB, but of worsening measured by the ADAS-Cog14 in patients with mild AD. It is notable that the cNTB and ADAS-cog14 did not provide consistent results. Although both assess cognition, the cNTB is slanted toward executive function, working and episodic memory, while the ADAS-cog is weighted toward orientation, recall, and language. In addition, the cNTB also considers response time, and drugs that enhance wakefulness or alertness, may lead to improvement in this measure. There is now compelling evidence for the association between shorter sleep duration and poor sleep quality to greater beta amyloid burden and higher risk for AD (21-23). In line with the reports on association between progressive cognitive deterioration in healthy and in AD patients with parallel deterioration in sleep quality and quantity, drugs that enhance wakefulness may result in worsening the dementia as reflected by the worsening in ADAS-cog14 and PSQI in the carriers of the polymorphism cluster. Whereas piromelatine at similar doses has shown efficacy in improving sleep maintenance in patients with insomnia, the 6SNP polymorphism carriers seem to have a paradoxical response to the drug by apparently enhancing alertness. Worsening in ADAS-cog14 in the carriers may be explained by this paradoxical alerting effect.
Thus far, the intergenic 2:107,510,000-107,540,000 range has not been described in relation to any disease. However, copy number variants in chromosome 2q12 region encompassing the 2:107,510,000-107,540,000 range are associated with schizophrenia. Rare copy number variants (deletions or duplications; CNVs) in Chromosome 2 region 2q12.2 (2: 106992995-108507424 in GRCh37) in vicinity of or encompassing the 2:107,510,000-107,540,000 locus) were reported in three of 1656 (0.18%) people with schizophrenia and in one of 4036 (0.02%) healthy controls (24) and suggested as a schizophrenia susceptibility risk factor, but not formally associated with the disease nor with symptomatic AD. None of the genes in the deleted segments were implicated in schizophrenia. The 6 single-nucleotide polymorphisms of the 2:107,510,000-107,540,000 range are intergenic between the ST6GAL2 gene: chr2:107,418,056-107,503,564 (downstream of the 2q12 polymorphism cluster) and the PPP1R2P5 gene: chr2:107,557,336-107,557,938 (upstream of the 2q12 polymorphism cluster). These two genes are not associated with AD or sleep quality. None of these publications disclosed the high proximity the 2q12 SNPs and their high frequency (ca. 27%) among early symptomatic AD patients.
We could not predict the functional impact of the 2q12 polymorphism cluster on gene expression and the mechanism by which it may affect response to piromelatine treatment. A SNP present within a coding sequence leading to an amino acid change (i.e., a non-synonymous SNP) can modify the protein’s activity. In our case the 2q12 polymorphism cluster is synonymous and may alter programmed translational speed and affect expression and function of an unknown encoded protein including 5-HT receptors and metabolic pathways, some known to be associated with wakefulness (25). As piromelatine is a 5HT-1A agonist, the link between the polymorphism and 5HT-1A pathways remains to be investigated. Thus, the inclusion of mild AD patients who are carriers of the (2:107,510,000-107,540,000) 5-6 SNPs polymorphism cluster in clinical trials may mask beneficial effects of piromelatine on non-carriers.
This study suggests that piromelatine, especially in higher doses, may be particularly effective for mild AD patients who do not carry this 2q12 polymorphism cluster. These findings warrant further investigation in a larger, prospective clinical trial in Alzheimer disease patients who do not carry the 2q12 polymorphism cluster.
Limitations of our study include the small number of individuals as compared with many GWAS studies and the post hoc approach; and, therefore, the limited power to detect variants associated with piromelatine response.
In summary, we identified a polymorphism cluster that appears associated with greater, but paradoxical, response to piromelatine treatment. If confirmed, then AD patients with SNPs associated with low likelihood of response to piromelatine may benefit from different therapies.
Conflict of Interest/Disclosure Statement: Nava Zisapel is the founder and CSO of Neurim Pharmaceuticals, the sponsor of the ReCognition study. Dr. Schneider reports personal fees from Neurim, Ltd, during the conduct of the study; grants and personal fees from Eli Lilly, personal fees from Boehringer Ingelheim, grants and personal fees from Merck, personal fees from Cognition, grants from Eisai, personal fees from Takeda, personal fees from vTv, grants and personal fees from Roche/Genentech, grants from Biogen, grants from Novartis, grants from Biohaven, grants from Washington Univ/ NIA DIAN-TU, personal fees from Samus, personal fees from IBC, personal fees from Cortexyme, personal fees from Alpha Cognition, personal fees from Novo-Nordisk, outside the submitted work.
ClinicalTrials.gov Identifier: NCT02615002.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. FDA Aducanumab (marketed as Aduhelm) Information. [https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/aducanumab-marketed-aduhelm-information]
2. Benca RM, Teodorescu M: Sleep physiology and disorders in aging and dementia. Handb Clin Neurol 2019, 167:477-493.
3. Nous A, Engelborghs S, Smolders I: Melatonin levels in the Alzheimer’s disease continuum: a systematic review. Alzheimers Res Ther 2021, 13(1):52.
4. Wade AG, Farmer M, Harari G, Fund N, Laudon M, Nir T, Frydman-Marom A, Zisapel N: Add-on prolonged-release melatonin for cognitive function and sleep in mild to moderate Alzheimer’s disease: a 6-month, randomized, placebo-controlled, multicenter trial. Clin Interv Aging 2014, 9:947-961.
5. Fava M, Targum SD, Nierenberg AA, Bleicher LS, Carter TA, Wedel PC, Hen R, Gage FH, Barlow C: An exploratory study of combination buspirone and melatonin SR in major depressive disorder (MDD): a possible role for neurogenesis in drug discovery. J Psychiatr Res 2012, 46(12):1553-1563.
6. NIH: Sleep Laboratory Study to Investigate the Safety and Efficacy of Neu-P11 in Primary Insomnia Patients NCT01489969. 2018.
7. Grandy J: Melatonin: Therapeutic Intervention in Mild Cognitive Impairment and Alzheimer Disease. . J Neurol Neurophysiol 2013, 4(2):148.
8. Merica H, Blois R, Gaillard JM: Spectral characteristics of sleep EEG in chronic insomnia. Eur J Neurosci 1998, 10(5):1826-1834.
9. NIH: Safety and Efficacy of Piromelatine in Mild Alzheimer’s Disease Patients (ReCOGNITION) NCT02615002. 2020.
10. Harrison J, Minassian S, Jenkins L, Black R, Koller M, Grundman M: A neuropsychological test battery for use in Alzheimer disease clinical trials. ;(9):). Arch Neurol 2007, 64(9):1323-1329.
11. Mohs RC KD, Petersen RC, et al. : Development of cognitive instruments for use in clinical trials of antidementia drugs: additions to the Alzheimer’s Disease Assessment Scale that broaden its scope. . Alzheimer Dis Assoc Disord 1997, 11:S13-S21.
12. Buysse D, Reynolds Cr, Monk T, Berman S, Kupfer D: The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. May;. Psychiatry Res 1989 28(2):193-213.
13. Fernandes BS WL, Steiner J, Leboyer M, Carvalho AF, Berk M. : The new field of ‘precision psychiatry’ BMC Med 2017, 15(1):80.
14. Myers A, Nemeroff C: New Vistas in the Management of Treatment-Refractory Psychiatric Disorders: Genomics and Personalized Medicine. Focus 2010, 8:525-535.
15. Schneider L, Olin J, Doody R, Clark C, Morris J, Reisberg B, Schmitt F, Grundman M, Thomas R, Ferris S: Validity and reliability of the Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change. The Alzheimer’s Disease Cooperative Study. . Alzheimer Disease & Associated Disorders 1997, 11:S22-32.
16. Galasko D BD, Sano M, Ernesto C, Thomas R, Grundman M, Ferris S. : An inventory to assess activities of daily living for clinical trials in Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. . Alzheimer Dis Assoc Disord 1997, 11:S33-39.
17. Variant Quality Score Recalibration (VQSR) [https://gatk.broadinstitute.org/hc/en-us/articles/360035531612-Variant-Quality-Score-Recalibration-VQSR-]
18. Norman GR, Sloan JA, Wyrwich KW: Interpretation of changes in health-related quality of life: the remarkable universality of half a standard deviation. Med Care 2003, 41(5):582-592.
19. Kennedy R, Cutter G, Schneider L: Effect of APOE genotype status on targeted clinical trials outcomes and efficiency in dementia and mild cognitive impairment resulting from Alzheimer’s disease. . Alzheimer’s & dementia : the journal of the Alzheimer’s Association 2014,, 10(3):349-359.
20. Cacabelos R, Carril J, Cacabelos P, Teijido O, Goldgaber D: Pharmacogenomics of Alzheimer’s Disease: Genetic Determinants of Phenotypic Variation and Therapeutic Outcome. . J Genomic Med Pharmacogenomics, 2016, 1(2):151-209.
21. Spira A, Gamaldo A, An Y, Wu M, Simonsick E, Bilgel M, Zhou Y, Wong D, Ferrucci L, Resnick S: Self-reported sleep and β-amyloid deposition in community-dwelling older adults. JAMA Neurol 2013, 70(12):1537-1543.
22. Yaffe K FC, Hoang T. : Connections between sleep and cognition in older adults. . Lancet Neurol 2014, 13(10):1017-1028.
23. You JC JE, Cross DE, Lyon AC, Kang H, Newberg AB, Lippa CF. : Association of β-Amyloid Burden With Sleep Dysfunction and Cognitive Impairment in Elderly Individuals With Cognitive Disorders. . JAMA Netw Open Oct 2; 2019, 2:e1913383.
24. Magri C, Sacchetti E, Traversa M, Valsecchi P, Gardella R, Bonvicini C, Minelli A, Gennarelli M, Barlati S: New copy number variations in schizophrenia. PLoS One 2010, 5(10):e13422.
25. Dzoljic M, Ukponmwan O, Saxena P: 5-HT1-like receptor agonists enhance wakefulness. . Neuropharmacology 1992 31(7):623-633.