
F. Guerville1, P. De Souto Barreto1,2, I. Ader3, S. Andrieu2,4, L. Casteilla3, C. Dray5, N. Fazilleau1,6, S. Guyonnet1,2, D. Langin5,7, R. Liblau6, A. Parini5, P. Valet5, N. Vergnolle8, Y. Rolland1,2, B. Vellas1,2
1. Gerontopole of Toulouse, Institute of Ageing, Toulouse University Hospital (CHU Toulouse), F-31000 Toulouse, France; 2. Inserm UMR1027, University of Toulouse III, F-31000 Toulouse, France; 3. STROMALab, University of Toulouse, CNRS ERL5311, EFS, ENVT, Inserm U1031, Université Paul Sabatier, F-31432 Toulouse, France; 4. Department of Epidemiology and Public Health, CHU Toulouse, Toulouse, France; 5. Institut des Maladies Métaboliques et Cardiovasculaires, Inserm UMR1048, University of Toulouse III, F-31000 Toulouse, France; 6. Centre de Physiopathologie de Toulouse Purpan, Inserm UMR1043, CNRS UMR5282, University of Toulouse III, F-31000 Toulouse, France; 7. Department of Medical Biochemistry, Toulouse University Hospital (CHU Toulouse), F-31000 Toulouse, France; 8. Institut de Recherche en Santé Digestive, Inserm UMR1220, INRA UMR1416, University of Toulouse III, F-31000 Toulouse, France
Corresponding Author: Florent Guerville, Institut du Vieillissement, Gérontopôle de Toulouse, 37 allée Jules Guesde, 31000 Toulouse, France. Email: florent.guerville@chu-bordeaux.fr, Tel: 0033561145664. Fax: 0033561145640
J Prev Alz Dis
Published online December 16, 2019, http://dx.doi.org/10.14283/jpad.2019.50
Abstract
The Geroscience aims at a better understanding of the biological processes of aging, to prevent and/or delay the onset of chronic diseases and disability as well as to reduce the severity of these adverse clinical outcomes. Geroscience thus open up new perspectives of care to live a healthy aging, that is to say without dependency. To date, life expectancy in healthy aging is not increasing as fast as lifespan. The identification of biomarkers of aging is critical to predict adverse outcomes during aging, to implement interventions to reduce them, and to monitor the response to these interventions. In this narrative review, we gathered information about biomarkers of aging under the perspective of Geroscience. Based on the current literature, for each hallmark of biological aging, we proposed a putative biomarker of healthy aging, chosen for their association with mortality, age-related chronic diseases, frailty and/or functional loss. We also discussed how they could be validated as useful predictive biomarkers.
Key words: Biomarkers, biological age, healthy aging, frailty, geroscience.
Geroscience from lifespan to healthy aging
The emerging field of Geroscience aims at a better understanding of the biological processes of aging, in order to reduce the burden of age-related diseases, slow functional decline and promote healthy aging (1–3). Human life expectancy remarkably increased worldwide during the past century and this rise is projected to continue (4). This is accompanied by an increasing prevalence of chronic diseases, including diabetes, cardio-vascular, neurodegenerative or kidney diseases and cancer, which share age as a common strong risk factor (5). Another critical challenge to societies is the amount of disability generated by these changes (6). Thus, healthy aging, the portion of life free of major chronic disease and disability, is not increasing to the same extent as lifespan. Indeed, recent increase in life expectancy is thought to be mainly due to prolonged survival with chronic disease(s) and/or disability, rather than to healthy aging. Therefore, the compression of comorbidity (7), i.e. delaying chronic diseases as close as possible to natural death, has become a major goal to achieve. Another major obstacle to increase healthy aging is the decline in physiological (including physical and cognitive) functions that occurs with aging, with a strong negative impact on quality of life, independency and survival. Functional decline may be a consequence of chronic diseases, but may also occur independently of them (8). Thus, delaying, minimizing or even preventing functional decline are also major aims for Geroscience.
The need for biomarkers of healthy aging
“If you cannot measure it, you cannot improve it”, stated William Thomson, the great Irish physicist better known as Lord Kelvin. Following this principle, the identification of biomarkers of healthy aging is critical to predict adverse outcomes in late life, to implement interventions aiming at increasing healthy aging, and monitor the response to these interventions.
We especially need biological biomarkers that could capture the inter-individual variability of biological processes of aging before it becomes clinically detectable. Indeed, interventions to promote healthy aging might be more effective in people at risk for functional decline than in those already engaged in the disability process (9,10) Targeting proper interventions on people at risk would also reduce unnecessary health care costs on healthy individuals. For clinical trials, risk stratification based on biology would also be helpful to reduce sample size and study time period, through selection of participants with a high risk of clinical adverse outcomes. Furthermore, research on the biology of aging is probably more likely to identify shared molecular and cellular mechanisms of multiple age-related diseases and functional loss, thereby paving the way to targeted and personalized interventions (1,2,11).
One of the difficulties in identifying biomarkers of aging is that there is no consensus about an operational definition of biological aging. The American Federation of Aging Research (AFAR) defined 3 criteria that a biomarker of aging should ideally meet: mark the individual stage of aging and predict mortality better than chronological age; monitor aging in a range of systems and not the effects of diseases; and allow longitudinal non-invasive tracking in animals and humans (12). Then, which event(s) should be predicted by an ideal biomarker or set of biomarkers? Death is obviously a significant outcome, but can be preceded by a long period of multi-morbidity and disability, so time-to-death per se is not a relevant outcome for a biomarker of healthy aging. Age-related diseases are to be considered but this disease-centered approach may focus research on a specific organ or on one limited physiological system. Frailty, conceptually defined as an age-associated state of increased vulnerability to stressors, can be considered as a clinical metric of biological aging. Indeed, operational definitions of frailty were widely validated as predictive of hospitalizations, disability, and death (13). There is also growing interest in measuring intrinsic capacity, a composite of all the physical and mental capacities of an individual (14), as a key determinant of functional ability.
Bibliography methodological approch
For this narrative review, our search for putative biomarkers of healthy aging was based on the following criteria:
(I) In the absence of a consensual operational definition of biological aging, we searched for biomarkers associated with survival, several aging-related diseases, frailty and/or functional loss.
(II) Putative biomarkers should have been studied in humans. Whenever available, animal data were also considered.
(III) To cover the main domains of aging biology, we chose to report at least one putative biomarker of healthy aging for each of the nine hallmarks of aging proposed by Lopez-Otin et al. (15): genomic instability, telomere attrition, epigenetic changes, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem-cell exhaustion and altered intercellular communication. In Lopez-Otin et al.’s review, which was focused on mammals, each hallmark should ideally fulfill the following criteria: it should manifest during normal aging, its experimental aggravation should accelerate aging and its experimental amelioration should delay the normal aging process and thus increase healthy aging. Thus, there is causal evidence for the implication of these biological mechanisms in the aging process, and associated therapeutic potential.
(IV) In a feasibility purpose, we chose only non-invasive biomarkers.
(V) In a discovery purpose, we focused on the literature published after the review by Lopez-Otin et al. (2013).
The search was performed on PubMed in April 2019 using the following terms: “biomarker” and (“aging” or “frailty” or “functional decline” or “genomic instability” or “telomere attrition”, or “epigenetic changes” or “loss of proteostasis” or “deregulated nutrient sensing” or “mitochondrial dysfunction” or “cellular senescence” or “stem-cell exhaustion” or “inflammaging”). The nine latter keywords were selected based on the nine hallmarks of aging proposed by Lopez-Otin et al. (15).
We selected biomarkers that fulfilled criteria (I) (association with at least one cited outcome), (II), (III) and (IV). Criterion (V) was optional, because we found relevant literature published before 2013.
Putative biomarkers of healthy aging
The results of our search are summarized in the Table. Only blood-based biomarkers met our selection criteria. We present below putative biomarkers for each hallmark of aging.
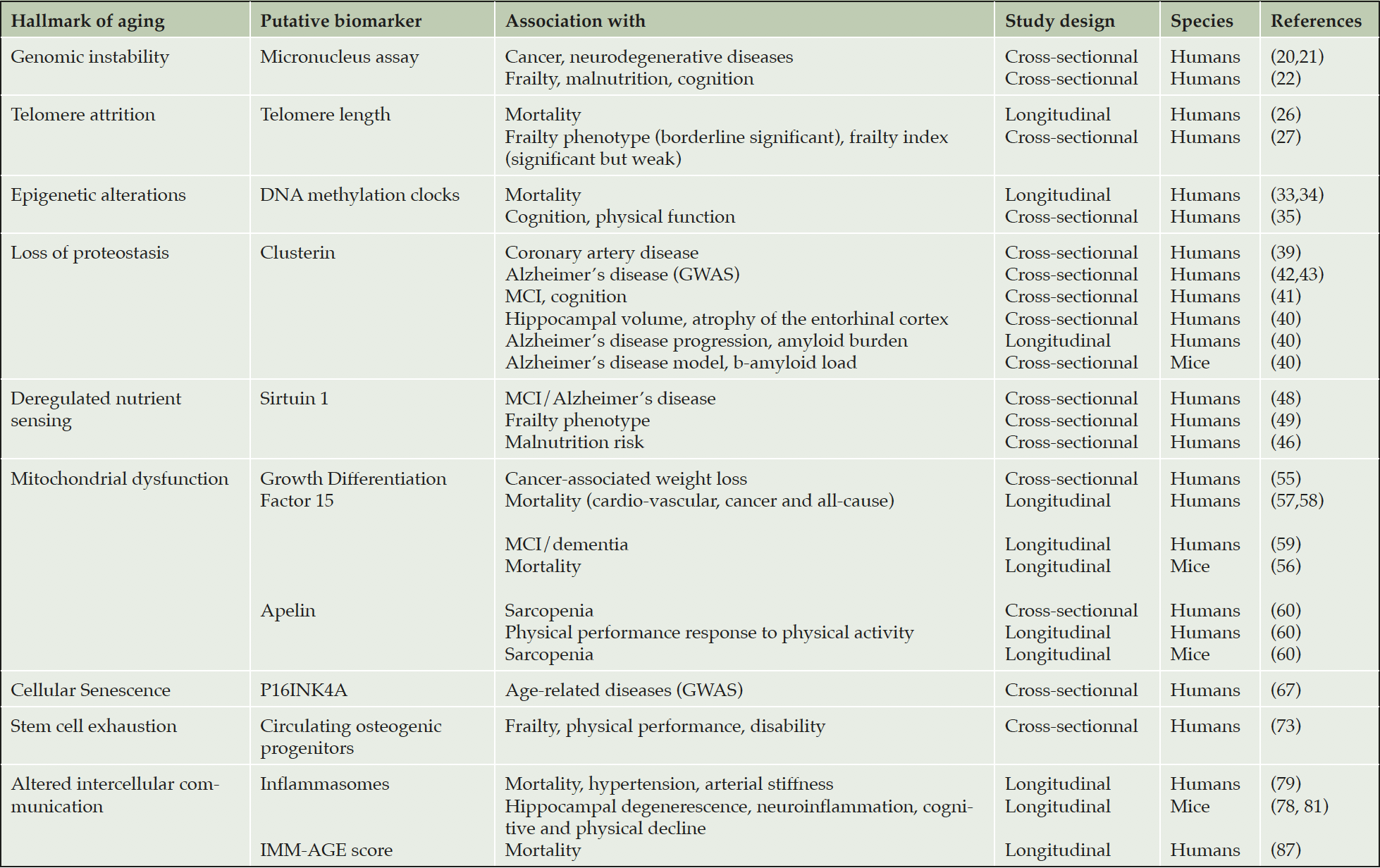
Table 1. Putative biomarkers of healthy aging
MCI, mild cognitive impairment. GWAS, genome-wide association studies
Genomic instability: Micronucleus assay
Genetic damage accumulates with aging, due to extrinsic and intrinsic factors, and genomic instability results from the imbalance between DNA damage and repair (16,17). Chromosome damage can be assessed with the micronucleus assay, which measures chromosome loss and breakage (18). Micronuclei are formed from chromosome fragments or whole chromosomes left out during cell division. From a minimum of 2000 cells, the percentage of micro-nucleated cells is measured via automatic microscope scoring and reviewed by an experienced scorer (19). Due to their non-invasive availability, peripheral blood and exfoliated buccal cells are the preferred material for this assay. The percentage of micro-nucleated cells increases with age, cancer, neurodegenerative diseases, tobacco use, and decreases with fruit consumption (20,21).
In 257 persons aged 65 and older from Galicia (Spain), Sanchez-Flores et al. recently reported a cross-sectional association between frailty and the micronucleus assay performed in peripheral blood lymphocytes (22). Interestingly, in this study, a higher micronucleus frequency was associated with 4 over 5 criteria of Fried frailty phenotype (except unintentional weight loss), with malnutrition or risk of malnutrition according to the Mini Nutritional Assessment score and with cognitive impairment according to the Mini-Mental Status Examination score. Longitudinal studies are required to validate the micronucleus assay as a healthy aging biomarker. In animals, the micronucleus assay has been widely used as a genotoxicity test (23), but not as a biomarker of healthy aging.
Telomere attrition
Some chromosome regions are particularly susceptible to age-related damage: telomeres are repetitive DNA sequences capping chromosomes, which shortens every time cells divide. It is probably the most studied hallmark of aging, with more than 8000 publications referenced in PubMed to date. The two main historical methods used to measure telomere length are the Southern blot (measuring the size of enzymatically-cleaved telomere fragments (24) and the quantitative polymerase chain reaction (qPCR), which reports a telomere/single copy gene signals ratio (25).
In a recent meta-analysis of twenty-five studies (n=121749, 21763 deaths) telomere attrition was predictive of all-cause mortality: subjects with telomere length in the lowest quartile had a 26% (95% CI 15-38%) higher hazard of death (26). The relation with frailty is less clear: in a meta-analysis of nine studies (n=10079 older subjects), Araujo-Carvalho et al. reported a borderline positive association between telomere attrition and Fried frailty phenotype (standard mean difference -0.56, 95% IC -1.12 to 0.00) and a statistically significant but weak positive association between telomere attrition and frailty index (standard mean difference 0.06; 95% IC -0.10 to -0.01) (27). The authors concluded that telomere length may not be a meaningful biomarker for frailty.
Nevertheless, attrition is not the only telomere modification observed during aging. Indeed, data from human and mice suggest a contribution of telomere damage to lung and cardiomyocyte aging, independently of telomere length (28,29). Interestingly, these works highlight molecular links between several hallmarks of aging: telomere damage is driven by mitochondrial dysfunction (through reactive oxygen species) and contributes to cellular senescence. Further investigations are needed to assess if telomere damage, detected noninvasively, could predict health outcomes during aging.
Epigenetic alterations: DNA methylation clocks
Changes in DNA sequence are not the only age-related genomic alterations. Epigenetic modifications such as DNA methylation, histone modification, chromatin remodeling, that influence gene expression, are also features of aging (15). Among them, changes in methylation of CpG islets are major regulators of gene expression. Based on these changes, relatively constant between individuals, several groups identified “DNA methylation clocks” that accurately predicts the chronological age of the donor (30,31). The clock by Hannum et al., developed from whole blood DNA and measuring methylation fraction of 25000 CpG islets, has a correlation coefficient with age >0.9 and an average error in age prediction <5 years. Nevertheless, a healthy aging biomarker should measure biological age rather than chronological age. Interestingly, DNA methylation clocks are considered as hybrid measurement, involving both chronological and biological elements (32).
Indeed, biological age may be reflected by the difference between true chronological age and DNA methylation age (i.e. age predicted by a DNA methylation clock). In four cohorts of older persons from Scotland and USA (n=4658), this difference (Δage) was found predictive of mortality: a 5-year Δage was associated with a 16% increase in mortality risk, independently of age, education, social class and comorbidity (33). A simpler score, based on methylation of only 10 CpG sites, was also reported predictive of all-cause, cardio-vascular and cancer mortality in two independent cohorts (34). Furthermore, in 1091 septuagenarians participating in one of the Scottish cohort cited above (LBC1936), Marioni et al. reported a cross-sectional negative association between Δage, cognition (6 tests from the Wechsler Adult Intelligence Scale-III) and physical function (grip strength) (35). Nevertheless, neither Δage, nor its longitudinal change, were found predictive of cognitive or physical decline.
Loss of proteostasis: Clusterin
Intracellular protein homeostasis, or proteostasis, is maintained through several quality control mechanisms: protein refolding by chaperone proteins and degradation by the ubiquitin-proteasome system or lysosomal pathways (autophagy). Due to cellular stress increasing protein misfolding, and/or failure of quality control mechanisms, aggregation of misfolded proteins are features of aging and age-related diseases, such as Alzheimer’s (36, 37)
The soluble form of Clusterin (sCLU, also known as Apolipoprotein J) protects from protein aggregation and precipitation (38). Using different techniques, several groups reported associations between Clusterin and age-related diseases.
Using ultracentrifugation or gel filtration, Riwanto et al. isolated serum HDL-associated Clusterin and reported a decreased level in patients with coronary artery disease compared to healthy controls from Switzerland (39). In an elegant biomarker discovery report, Thambisetty et al. provided more insight about the potential role of Clusterin in Alzheimer’s disease (40). In the discovery phase of the study, proteomic analyses revealed a positive association between serum Clusterin and (a) hippocampal atrophy measured with MRI in 44 subjects with mild cognitive impairment (MCI) or mild to moderate AD from the KCL-ART study (London), and (b) disease progression speed according to the clinical ADAS-cog scale in 51 AD patients from the AddNeuroMed European cohort. In the validation phase, serum Clusterin (as measured by an ELISA technique) was positively associated with atrophy of the entorhinal cortex (as measured with MRI), severity of cognitive impairment and speed of progression in AD (as measured with MMSE before or after blood sampling) in 689 participants of the KCL-ART or the AddNeuroMed study. Furthermore, in 60 non-demented participants of the Baltimore Longitudinal Study of Aging, serum Clusterin was positively associated with fibrillar amyloid burden in the entorhinal cortex, as measured with PET imaging 10 years after blood sampling. Finally, in a mouse model of AD, serum Clusterin was higher than in wild-type mice, cortical plaques contained both Amyloid-β protein and Clusterin, and the cortical loads of the 2 proteins were highly positively correlated.
Using an APO multiplex bead fluorescence immunoassay technique in 664 participants (257 with MCI) of the Sydney Memory and Aging Study, Song et al. reported higher levels of serum Clusterin/APOJ in subjects with MCI, and a negative correlation between APOJ levels and cognitive scores (41).
In two genome-wide association studies (>14000 people in France, Belgium, Italy, Finland and Spain and 16000 people in UK, Germany and USA), polymorphisms in the clusterin gene were found strongly associated with Alzheimer’s disease (AD), as was the well-established susceptibility locus APOE (42,43).
However, given the opposite direction of associations between serum Clusterin and coronary artery disease and AD, further research is needed to determine if Clusterin could be a biomarker of healthy aging.
Deregulated nutrient sensing: Sirtuins
Mammals’ somatotrophic axis comprises the growth hormone and the insulin-like growth factor (IGF-1), which shares downstream intracellular pathway with insulin, thereby signaling nutrient abundance and anabolism. Decline in this axis is one of the major features of metabolic aging (44). Besides the insulin and IGF-1 signaling pathway, sirtuins are other nutrient sensors with an opposite effect: they signal nutrient scarcity and catabolism. Thus, activation of sirtuins mimics calorie restriction and improves lifespan and health in animals (45).
Performing RT-PCR on whole blood cells from 350 community-dwellers participating to the Toledo Study for Healthy Aging, El Assar et al. recently tested the association between the transcription of 21 genes involved in response to stress and malnutrition risk assessed with the Mini-Nutritional Assessment score. The expression of sirt1, coding for sirtuin-1, was negatively associated with malnutrition risk, independently of age, comorbidity, frailty and diet (46). No associations were found between other genes and malnutrition risk. In addition, sirt1 plays a central role in survival and regeneration of skeletal muscle cells, as reviewed by Sharples et al. (47).
Sirtuin-1 was originally described as a nuclear protein, but was more recently reported detectable in human serum using ELISA, surface plasmon resonance and Western blot (48). In this first study, lower serum sirtuin-1 levels were found in healthy older (n=22) individuals and in MCI (n=9) or AD (n=40) patients than in young controls (n=22). In 200 Indian outpatients of a Geriatric Medicine Department, the same group reported lower serums sirtuins 1, 2 and 3 (as measured with surface plasmon resonance and Western blot) as independently associated with Fried frailty phenotype. A better diagnostic accuracy was found for sirtuin-1 (receiver operating characteristic’s area under curve = 0.9) (49). Despite external replication of the detection of sirtuin-1 in human serum (50), it is still unknown how and why this nuclear protein is released in the extracellular compartment.
Mitochondrial dysfunction: Growth Differentiation Factor 15 and Apelin
Human aging is generally linked to a progressive mitochondrial dysfunction (51). Among the important parameters involved in this dysfunction, the decrease in the efficacy of the respiratory chain observed in aging is characterized by increased reactive oxygen species (ROS) production, mitochondrial integrity defects and reduced mitochondrial biogenesis (controlled, among others, by sirtuins). Nevertheless, higher mitochondrial oxidative stress increases lifespan in rodents. These paradoxical effects of ROS on aging can be harmonized if their production is seen as a stress-compensatory mechanism to maintain survival, which becomes detrimental if excessive and sustained (52).
Growth differentiation factor 15 (GDF-15) is a stress-induced cytokine and member of the transforming growth factor β superfamily. GDF-15 has emerged as a biomarker of cellular stress than can be produced by a number of organs such as lung, kidney and liver (53). It is also considered as a diagnostic marker for inherited mitochondrial diseases, and potentially as a marker of mitochondrial dysfunction (54). GDF-15 has negative effects on appetite and weight in mice and is associated with weight loss in patients with cancer (55). Furthermore, its overexpression increases lifespan in mice, especially on a high-fat diet (56). In two Swedish cohorts (n=1200), higher GDF-15 serum levels (measured by ELISA) was associated with cardio-vascular, cancer and all-cause 5- and 12-year mortality, independently of telomere length, IL-6 and CRP (57). Measured by an immunoradiometric assay in frozen plasma in 1000 septuagenarians participants of the PIVUS study (Sweden), longitudinal increase in GDF-15 levels was associated with a 4-fold increase in the 5-years mortality hazard (58). Finally, in 1037 non demented community-dwellers >70 yo participants to the Sydney Memory and Aging Study, higher serum GDF-15 (measured by ELISA) was associated with MCI/dementia incidence, independently of cardiovascular comorbidity, APOE genotype and inflammation parameters (59). Even if expression and secretion of GDF-15 are increased in response to deterioration of energy metabolism in a cellular model of mitochondrial disease (54), the physiological link between GDF-15 and mitochondrial dysfunction, especially during aging, remains to be determined.
Recent findings suggest that apelin, an exercise-induced myokine, may also be considered as a putative biomarker of healthy aging related to mitochondrial dysfunction (60). Among 61 participants of the French MAPT study aged 70 and older, baseline serum apelin (measured with ELISA) was positively associated with muscle mass (measured using dual energy X-ray absorptiometry), independently of age, sex and BMI. Moreover, increase of serum apelin over 6 month was positively correlated to physical function improvement (SPPB score) in 34 participants >70 yo of the physical activity LIFE-P trial. In the same work, apelin production by muscle declined with aging in mice while sarcopenia was exacerbated in apelin-deficient mice and was reversed by apelin supplementation or overexpression. In those experiments, apelin enhanced muscle function through mitochondriogenesis, but also other pathways related to hallmarks of aging: autophagy, inflammation and muscle stem cells. It remains to be determined whether apelin could predict other outcomes than sarcopenia and response to exercise, such as Alzheimer’s disease (61), and could be considered as a broader biomarker of healthy aging.
Cellular senescence: p16Ink4A
Cellular senescence is a state of stable arrest of the cell cycle coupled to phenotypic changes, including the production of several molecules (especially matrix metalloproteases and pro-inflammatory cytokines) collectively known as the senescence-associated secretory phenotype (SASP) (62). The SASP mediates senescence spreading to adjacent cells, inflammation, and tissue dysfunction. Seen as a compensatory mechanism aimed at avoiding proliferation of damaged cells, cellular senescence is induced by age-associated stimuli: telomere attrition, DNA damage and excessive mitogenic signaling, particularly by the p16Ink4a tumor suppressor protein, upon epigenetic de-repression of the ink4/ark locus (63).
p16Ink4A positively correlates with age in various tissues in mice and in human skin (64,65). Measured by RT-PCR in peripheral blood T lymphocytes from 170 donors of 2 independent US cohorts, the transcription of p16Ink4a was positively associated with age, tobacco use and physical inactivity (66). Moreover, in a meta-analysis of 372 GWAS studies aiming at identifying susceptibility polymorphisms for age-associated diseases, the ink4/ark locus was linked to the highest number of diseases, including Alzheimer’s, cardio-vascular diseases, cancer and type 2 diabetes (67).
To our knowledge, an association between a marker of cellular senescence and functional loss, frailty or aging phenotype has not yet been reported. As recently suggested, a set of biomarkers would be more efficient to capture the accumulation of senescent cells during aging (68). Given the central role of the SASP in consequences of cellular senescence, a systemic measurement of key components of the SASP in an available sample (like blood) would be, if associated with functional loss or an aging phenotype, an interesting biomarker of healthy aging. In view of the association between senescent cells accumulation and several age-associated diseases, removing senescent cells from tissues is a promising pharmacological target (69).
Stem cell exhaustion: Circulating osteogenic progenitors
The repair and regenerative potential of many tissues declines with aging, due to functional attrition in several stem cell compartments (e.g. hematopoietic, neural, mesenchymal and intestinal epithelial stem cells, as well as satellite cells in muscles). Adult stem cells are present in every tissues and organs after development and regenerate damaged tissues throughout life. During aging, the function of stem cells decline (70). Stem cell exhaustion is seen as an integrative consequence of several hallmarks of aging described above, including DNA damage, epigenetic alterations, telomere shortening, cellular senescence and mitochondrial dysfunction (15).
However, stem cell exhaustion is difficult to measure non-invasively before the onset of its clinical consequences, such as anemia and other cytopenias for hematopoietic stem cells, but also sarcopenia for muscle stem cells/satellite cells, and decreased intestinal function for intestinal epithelial stem cells. So far, data is scarce on potential biomarkers for this hallmark of aging. Circulating osteogenic progenitors (COP) cells were proposed as a surrogate marker of the mesenchymal stem cell population within the bone marrow (71). Their ability to differentiate, not only into bone, but other mesenchymal tissues, including muscle, offers perspectives in regenerative medicine for musculoskeletal diseases (72). In 77 participants of the Nepean osteoporosis and Frailty study older than 65 yo, the proportion of COP cells among peripheral blood mononuclear cells was measured using flow cytometry, as double positive cells for CD45 (an hematopoietic marker) and osteocalcin (a marker of bone formation). COP cell percentage was inversely correlated with age. Lower COP cell percentages were associated with frailty, lower physical performance (measured by grip strength and gait speed) and disability, independently of age and comorbidity (73). Nevertheless, there is currently no consensual phenotype to specifically identify these cells in blood (72) and no longitudinal associations with lifespan or healthy aging have so far been reported.
Altered intercellular communication: Inflammasomes and IMM-AGE score
Aging is associated with changes in communications between cells, mainly driven by a chronic low-grade systemic inflammation named inflammaging (15,74). This inflammation is seen as a consequence of several hallmarks of aging described above, including cellular senescence (through the SASP) and loss of proteostasis, because misfolded proteins constitute a danger signal that triggers the innate immune response (75). A large body of literature links inflammaging to age-associated diseases, functional decline, and frailty (76).
One of the major pathways of inflammaging is the inflammasome pathway. Firstly described in innate immune cells (77), the inflammasome describes a complex system of intracellular proteins that assembly upon detection of stress/danger signals and trigger maturation and release of pro-inflammatory cytokines (namely interleukin-1β and interleukin-18). Mouse models lacking the NLRP3 inflammasome exhibit less inflammaging, glucose intolerance, hippocampal degenerescence, neuroinflammation, cognitive and physical decline (78). In participants of the Stanford-Ellison cohort aged 60 to >90 yo, inflammasome activation (measured by nlcrc4 and nlrc5 genes expression in whole blood cells and interleukin-1β production) was positively associated with hypertension and arterial stiffness and negatively associated with personal and familial longevity (79). As cholesterol crystals and β-amyloid proteins can trigger assembly of inflammasome complexes, this pathway is involved in atherosclerosis lesion progression (80) and neuro-inflammation in AD (81,82). Thus, inflammasome inhibitors are promising drugs in age-related diseases (83–85).
Beyond inflammaging, immunosenescence encompasses quantitative and functional changes of multiple actors of both the innate and adaptive arms of the immune system (86). Immunosenescence may aggravate the aging process related to hallmarks of aging described above, notably because of the failure to eliminate pathogens, but also pre-malignant cells, senescent cells and misfolded proteins (15,75). Using an integrative and longitudinal “multi-omics” approach from peripheral blood, Alpert et al. recently captured the immune system trajectories in 135 healthy older individuals (87). Moreover, they derived a simplified “IMM-AGE” score based on baseline expression of 57 immune genes that predicted all-cause mortality over 7 years, independently of cardio-vascular risk factors and disease, in >2000 participants of the Framingham Heart Study. Survival was far more significantly associated with the IMM-AGE score than with the DNA methylation age in the same population. This work provides major contributions, especially regarding inter-individual variability of immunosenescence trajectories and their prognostic value.
Conclusions and perspectives
To date, no (set of) biomarker(s) has been reported to fulfill ideal criteria for biomarkers of healthy aging: measuring aging in a range of systems, non-invasively in humans and animals, predicting mortality, age-related diseases and loss of functions. Nevertheless, we report here several putative blood biomarkers that were shown predictive of mortality and/or associated with age-related chronic diseases and/or functional decline. Some of them (e.g. DNA methylation clocks) were externally validated, but most of them were not. Above all, associations between these putative biomarkers and frailty or loss of functions are mostly cross-sectional. Therefore, there is a major need for longitudinal studies with repeated measures of physical and mental functions in participants of a wide range of age and health status. Especially, cohorts including middle-aged persons would allow the identification of early biomarkers of healthy aging, whereas such biomarkers could be missed in studies focusing on older people, due to selection bias. Longitudinal assessment of putative biomarkers would also allow studying their dynamic. This would certainly provide more insight in the biological processes of aging and their heterogeneity across individuals (87).
Giving the complexity of the aging process, the probability that a single biomarker will ever meet those ideal criteria seems very low. At the opposite, the availability of “omics” approaches now allows hypothesis-free identification of potential biomarkers, not only among genes, transcripts and proteins, but also among non-coding RNA and metabolites (88–90). How to integrate, with a physiological perspective, hypothesis-driven approaches focused on a single biological pathway and multi-omics approaches is probably one of the major challenge for future research on biomarkers of healthy aging. In that purpose, artificial intelligence has already been used to provide biological age prediction tools and its convergence with Geroscience is expected to grow (91). Another major, often underestimated, challenge in biomarker development is to meet the standards for widespread use in laboratory medicine (92,93).
We chose to focus on the nine hallmarks of aging proposed by Lopez-Otin et al. (15), but new hallmarks may emerge. For example, all the studies described above concentrated their efforts on investigating host biomarkers of healthy aging. A growing field related to the identification of microbial strains (bacteria, virus, parasites, fungi) could soon add more candidates to the list of possible healthy aging biomarkers. Most work thus far has been rather descriptive. Gut microbiota dysbiosis has been associated with a number of diseases, but also with aging (94,95). Recent studies using turquoise killifish demonstrated that transfer of the gut microbiome from young to middle-aged killifish resulted in an increase in lifespan and a delayed behavioral decline, compared to fish that received the microbiota from middle-aged fish (96). The composition of human gut microbiome changes heavily from one individual to another and is also sensitive to many environmental factors, including diet or medication, which are important actors in aging individuals. Further experimental and clinical studies are needed to explore the role of microbiome (from the gut, but also potentially from the skin or the mouth) in aging, and to identify microbial healthy aging markers. This is a completely unexplored domain for the moment, which could well complement the search of host healthy aging biomarkers.
The use of animal models that age faster than humans and are more suitable for experimental modification of biologic pathways or life conditions is essential for biomarker discovery and validation. The five main model organisms used in aging-related research are budding yeast Saccharomyces cerevisiae, nematodes Caenorhabditis elegans, fruit flies Drosophila melanogaster, fishes Nothobranchius furzeri and laboratory mice Mus musculus. Numerous studies on these different animal models have identified several orthologous genes that modulate longevity in the same way over a long evolutionary distance (97). Of note, some vertebrates, like the African Killifish, age even faster than mice and are useful models to study the biology of aging (11). Each of these species has its limits and strengths as a model for human aging, and it is important to consider the way they look alike but also how they differ in physiology, longevity, and aging traits.
Efforts are needed to reduce differences between animal lab life and human real life. Indeed, lab animals usually have a homogeneous or even identical genetic background and live in pathogen-controlled conditions. Working on inbred or outbred mice to study biological processes of aging remains an open question (98). Their food intake has often been modified to influence the aging process (99), contrarily to their physical activity, despite possible effects on healthy aging (100). Finally, the development of preclinical models of frailty is an extremely important step in Geroscience research. Despite efforts in this direction in mice (101–103), translational studies on the mechanisms of aging in animals and humans have yet to be conducted.
Several non-pharmacological interventions, including diet and exercise, may influence lifespan and healthy aging through effects on several hallmarks of aging (99,104–106). It will be useful to take those parameters into account in human and animal studies designed to discover and validate biomarkers of healthy aging. A better understanding of the biology of aging also paves the way to potentially promising pharmacological interventions linked to several hallmarks of aging, including senolytics (69), inflammasome inhibitors (84), metformin (107), rapamycin (108), resveratrol (109,110) and mesenchymal stem cells (111). One could argue that improving the detection of frailty and functional loss and the implementation of personalized non-pharmacological interventions is more likely to increase healthy aging in populations than new or repurposed drugs (112). Biomarkers of healthy aging could nevertheless become useful as complementary tools to stratify the risk of functional loss and to monitor response to lifestyle interventions.
Conflict of interest/disclosures: The authors have no conflict of interest to disclose.
Funding: For this work, Florent Guerville received a grant (Bourse de Mobilité) from Bordeaux University Hospital.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. Seals DR, Justice JN, LaRocca TJ. Physiological geroscience: targeting function to increase healthspan and achieve optimal longevity: Translational physiology of ageing. J Physiol 2016;594:2001–24.
2 Partridge L, Deelen J, Slagboom PE. Facing up to the global challenges of ageing. Nature 2018;561:45–56.
3 Margolick JB, Ferrucci L. Accelerating aging research: how can we measure the rate of biologic aging? Exp Gerontol 2015;64:78–80.
4 Kontis V, Bennett JE, Mathers CD, et al. Future life expectancy in 35 industrialised countries: projections with a Bayesian model ensemble. The Lancet 2017;389:1323–35.
5 Niccoli T, Partridge L. Ageing as a risk factor for disease. Curr Biol 2012;22:R741–R752.
6 World Health Organizatioń. World report on ageing and health. 2015.
7 Fries JF. Aging, natural death, and the compression of morbidity. N Engl J Med 1980;303:130–5.
8 Seals DR, Melov S. Translational geroscience: emphasizing function to achieve optimal longevity. Aging 2014;6:718.
9 Gill TM, Baker DI, Gottschalk M, et al. A program to prevent functional decline in physically frail, elderly persons who live at home. N Engl J Med 2002;347:1068–74.
10 Beswick AD, Rees K, Dieppe P, et al. Complex interventions to improve physical function and maintain independent living in elderly people: a systematic review and meta-analysis. Lancet Lond Engl 2008;371:725–35.
11 Singh PP, Demmitt BA, Nath RD, et al. The genetics of aging: a vertebrate perspective. Cell 2019;177:200–20.
12 Johnson TE. Recent results: biomarkers of aging. Exp Gerontol 2006;41:1243–6.
13 Clegg A, Young J, Iliffe S, et al. Frailty in elderly people. The Lancet 2013;381:752–62.
14 Cesari M, Araujo de Carvalho I, Amuthavalli Thiyagarajan J, et al. Evidence for the domains supporting the construct of intrinsic capacity. J Gerontol Ser A 2018;73:1653–60.
15 López-Otín C, Blasco MA, Partridge L, et al. The Hallmarks of Aging. Cell 2013;153:1194–217.
16 Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med 2009;361:1475–85.
17 Moskalev AA, Shaposhnikov MV, Plyusnina EN, et al. The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res Rev 2013;12:661–84.
18 Fenech M. A lifetime passion for micronucleus cytome assays—reflections from Down Under. Mutat Res Mutat Res 2009;681:111–7.
19 Fenech M. Cytokinesis-block micronucleus cytome assay. Nat Protoc 2007;2:1084.
20 Bonassi S, Coskun E, Ceppi M, et al. The HUman MicroNucleus project on eXfoLiated buccal cells (HUMN(XL)): the role of life-style, host factors, occupational exposures, health status, and assay protocol. Mutat Res 2011;728:88–97.
21 Migliore L, Coppedè F, Fenech M, et al. Association of micronucleus frequency with neurodegenerative diseases. Mutagenesis 2011;26:85–92.
22 Sánchez-Flores M, Marcos-Pérez D, Lorenzo-López L, et al. Frailty syndrome and genomic instability in older adults: suitability of the cytome micronucleus assay as a diagnostic tool. J Gerontol Ser A 2018;73:864–72.
23 Hayashi M. The micronucleus test—most widely used in vivo genotoxicity test—. Genes Environ 2016;38:18.
24 Kimura M, Stone RC, Hunt SC, et al. Measurement of telomere length by the Southern blot analysis of terminal restriction fragment lengths. Nat Protoc 2010;5:1596.
25 Cawthon RM. Telomere measurement by quantitative PCR. Nucleic Acids Res 2002;30:e47–e47.
26 Wang Q, Zhan Y, Pedersen NL, et al. Telomere length and all-cause mortality: a meta-analysis. Ageing Res Rev 2018;48:11–20.
27 Araújo Carvalho AC, Tavares Mendes ML, da Silva Reis MC, et al. Telomere length and frailty in older adults-A systematic review and meta-analysis. Ageing Res Rev 2019;54:100914.
28 Birch J, Anderson RK, Correia-Melo C, et al. DNA damage response at telomeres contributes to lung aging and chronic obstructive pulmonary disease. Am J Physiol-Lung Cell Mol Physiol 2015;309:L1124–L1137.
29 Anderson R, Lagnado A, Maggiorani D, et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J 2019;38:e100492.
30 Hannum G, Guinney J, Zhao L, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 2013;49:359–67.
31 Horvath S. DNA methylation age of human tissues and cell types. Genome Biol 2013;14:3156.
32 Field AE, Robertson NA, Wang T, et al. DNA Methylation Clocks in Aging: Categories, Causes, and Consequences. Mol Cell 2018;71:882–95.
33 Marioni RE, Shah S, McRae AF, et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol 2015;16:25.
34 Zhang Y, Wilson R, Heiss J, et al. DNA methylation signatures in peripheral blood strongly predict all-cause mortality. Nat Commun 2017;8:14617.
35 Marioni RE, Shah S, McRae AF, et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int J Epidemiol 2015;44:1388–96.
36 Powers ET, Morimoto RI, Dillin A, et al. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem 2009;78:959–91.
37 Koga H, Kaushik S, Cuervo AM. Protein homeostasis and aging: The importance of exquisite quality control. Ageing Res Rev 2011;10:205–15.
38 Vanhooren V, Santos AN, Voutetakis K, et al. Protein modification and maintenance systems as biomarkers of ageing. Mech Ageing Dev 2015;151:71–84.
39 Riwanto M, Rohrer L, Roschitzki B, et al. Altered activation of endothelial anti-and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: role of high-density lipoprotein–proteome remodeling. Circulation 2013;127:891–904.
40 Thambisetty M, Simmons A, Velayudhan L, et al. Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch Gen Psychiatry 2010;67:739–48.
41 Song F, Poljak A, Crawford J, et al. Plasma apolipoprotein levels are associated with cognitive status and decline in a community cohort of older individuals. PloS One 2012;7:e34078.
42 Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 2009;41:1088.
43 Lambert J-C, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 2009;41:1094.
44 Barzilai N, Huffman DM, Muzumdar RH, et al. The critical role of metabolic pathways in aging. Diabetes 2012;61:1315–22.
45 Mitchell SJ, Martin-Montalvo A, Mercken EM, et al. The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep 2014;6:836–43.
46 El Assar M, Angulo J, Walter S, et al. Better Nutritional Status Is Positively Associated with mRNA Expression of SIRT1 in Community-Dwelling Older Adults in the Toledo Study for Healthy Aging. J Nutr 2018;148:1408–14.
47 Sharples AP, Hughes DC, Deane CS, et al. Longevity and skeletal muscle mass: the role of IGF signalling, the sirtuins, dietary restriction and protein intake. Aging Cell 2015;14:511–23.
48 Kumar R, Chaterjee P, Sharma PK, et al. Sirtuin1: a promising serum protein marker for early detection of Alzheimer’s disease. PloS One 2013;8:e61560.
49 Kumar R, Mohan N, Upadhyay AD, et al. Identification of serum sirtuins as novel noninvasive protein markers for frailty. Aging Cell 2014;13:975–80.
50 Yanagisawa S, Papaioannou AI, Papaporfyriou A, et al. Decreased serum sirtuin-1 in COPD. Chest 2017;152:343–52.
51 Wang Y, Hekimi S. Mitochondrial dysfunction and longevity in animals: Untangling the knot. Science 2015;350:1204–7.
52 Hekimi S, Lapointe J, Wen Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol 2011;21:569–76.
53 Patel S, Alvarez-Guaita A, Melvin A, et al. GDF15 Provides an Endocrine Signal of Nutritional Stress in Mice and Humans. Cell Metab 2019;29:707–718.e8.
54 Fujita Y, Taniguchi Y, Shinkai S, et al. Secreted growth differentiation factor 15 as a potential biomarker for mitochondrial dysfunctions in aging and age-related disorders. Geriatr Gerontol Int 2016;16:17–29.
55 Johnen H, Lin S, Kuffner T, et al. Tumor-induced anorexia and weight loss are mediated by the TGF-β superfamily cytokine MIC-1. Nat Med 2007;13:1333.
56 Wang X, Chrysovergis K, Kosak J, et al. hNAG-1 increases lifespan by regulating energy metabolism and insulin/IGF-1/mTOR signaling. Aging 2014;6:690–704.
57 Wiklund FE, Bennet AM, Magnusson PKE, et al. Macrophage inhibitory cytokine-1 (MIC-1/GDF15): a new marker of all-cause mortality. Aging Cell 2010;9:1057–64.
58 Eggers KM, Kempf T, Wallentin L, et al. Change in growth differentiation factor 15 concentrations over time independently predicts mortality in community-dwelling elderly individuals. Clin Chem 2013;59:1091–8.
59 Fuchs T, Trollor JN, Crawford J, et al. Macrophage inhibitory cytokine-1 is associated with cognitive impairment and predicts cognitive decline – the Sydney Memory and Aging Study. Aging Cell 2013;12:882–9.
60 Vinel C, Lukjanenko L, Batut A, et al. The exerkine apelin reverses age-associated sarcopenia. Nat Med 2018;24:1360–71.
61 Masoumi J, Abbasloui M, Parvan R, et al. Apelin, a promising target for Alzheimer disease prevention and treatment. Neuropeptides 2018;70:76–86.
62 Coppé J-P, Desprez P-Y, Krtolica A, et al. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010;5:99–118.
63 Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell 2007;130:223–33.
64 Krishnamurthy J, Torrice C, Ramsey MR, et al. Ink4a/Arf expression is a biomarker of aging. J Clin Invest 2004;114:1299–307.
65 Ressler S, Bartkova J, Niederegger H, et al. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell 2006;5:379–89.
66 Liu Y, Sanoff HK, Cho H, et al. Expression of p16INK4a in peripheral blood T-cells is a biomarker of human aging. Aging Cell 2009;8:439–48.
67 Jeck WR, Siebold AP, Sharpless NE. Review: a meta-analysis of GWAS and age-associated diseases. Aging Cell 2012;11:727–31.
68 Matjusaitis M, Chin G, Sarnoski EA, et al. Biomarkers to identify and isolate senescent cells. Ageing Res Rev 2016;29:1–12.
69 Van Deursen JM. Senolytic therapies for healthy longevity. Science 2019;364:636–7.
70 Schultz MB, Sinclair DA. When stem cells grow old: phenotypes and mechanisms of stem cell aging. Dev Camb Engl 2016;143:3–14.
71 Pignolo RJ, Kassem M. Circulating osteogenic cells: implications for injury, repair, and regeneration. J Bone Miner Res Off J Am Soc Bone Miner Res 2011;26:1685–93.
72 Feehan J, Nurgali K, Apostolopoulos V, et al. Circulating osteogenic precursor cells: Building bone from blood. EBioMedicine 2019;39:603–11.
73 Gunawardene P, Bermeo S, Vidal C, et al. Association between circulating osteogenic progenitor cells and disability and frailty in older persons: the Nepean osteoporosis and frailty study. J Gerontol Ser Biomed Sci Med Sci 2015;71:1124–30.
74 Franceschi C, Bonafè M. Centenarians as a model for healthy aging. Biochem Soc Trans 2003;31:457–61.
75 Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol 2015;14:388–405.
76 Ferrucci L, Fabbri E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol 2018:1.
77 Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002;10:417–26.
78 Youm Y-H, Grant RW, McCabe LR, et al. Canonical Nlrp3 Inflammasome Links Systemic Low-Grade Inflammation to Functional Decline in Aging. Cell Metab 2013;18:519–32.
79 Furman D, Chang J, Lartigue L, et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat Med 2017;23:174–84.
80 Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010;464:1357–61.
81 Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2012;493:674–8.
82 Venegas C, Kumar S, Franklin BS, et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017;552:355–61.
83 Hara Y, McKeehan N, Fillit HM. Translating the biology of aging into novel therapeutics for Alzheimer disease. Neurology 2019;92:84–93.
84 Swanson KV, Deng M, Ting JP-Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 2019;doi: 10.1038/s41577-019-0165-0.
85 Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 2017;377:1119–31.
86 Nikolich-Žugich J. The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol 2018;19:10–9.
87 Alpert A, Pickman Y, Leipold M, et al. A clinically meaningful metric of immune age derived from high-dimensional longitudinal monitoring. Nat Med 2019;25:487–95.
88 Peters MJ, Joehanes R, Pilling LC, et al. The transcriptional landscape of age in human peripheral blood. Nat Commun 2015;6:8570.
89 Hertel J, Friedrich N, Wittfeld K, et al. Measuring Biological Age via Metabonomics: The Metabolic Age Score. J Proteome Res 2016;15:400–10.
90 Zierer J, Menni C, Kastenmüller G, et al. Integration of “omics” data in aging research: from biomarkers to systems biology. Aging Cell 2015;14:933–44.
91 Zhavoronkov A, Mamoshina P. Deep Aging Clocks: The Emergence of AI-Based Biomarkers of Aging and Longevity. Trends Pharmacol Sci 2019;40:546–9.
92 Taguchi A, Hanash SM. Unleashing the power of proteomics to develop blood-based cancer markers. Clin Chem 2013;59:119–26.
93 Aleksandrova K, Mozaffarian D, Pischon T. Addressing the Perfect Storm: Biomarkers in Obesity and Pathophysiology of Cardiometabolic Risk. Clin Chem 2018;64:142–53.
94 Kundu P, Blacher E, Elinav E, et al. Our Gut Microbiome: The Evolving Inner Self. Cell 2017;171:1481–93.
95 Schmidt TSB, Raes J, Bork P. The Human Gut Microbiome: From Association to Modulation. Cell 2018;172:1198–215.
96 Smith P, Willemsen D, Popkes M, et al. Regulation of life span by the gut microbiota in the short-lived African turquoise killifish. eLife 2017;doi: 10.7554/eLife.27014.
97 Bitto A, Wang AM, Bennett CF, et al. Biochemical Genetic Pathways that Modulate Aging in Multiple Species. Cold Spring Harb Perspect Med 2015;doi: 10.1101/cshperspect.a025114.
98 Tuttle AH, Philip VM, Chesler EJ, et al. Comparing phenotypic variation between inbred and outbred mice. Nat Methods 2018;15:994–6.
99 Fontana L, Partridge L. Promoting health and longevity through diet: from model organisms to humans. Cell 2015;161:106–18.
100 Garcia-Valles R, Gomez-Cabrera MC, Rodriguez-Mañas L, et al. Life-long spontaneous exercise does not prolong lifespan but improves health span in mice. Longev Heal 2013;2:14.
101 Parks RJ, Fares E, MacDonald JK, et al. A procedure for creating a frailty index based on deficit accumulation in aging mice. J Gerontol Ser Biomed Sci Med Sci 2011;67:217–27.
102 Whitehead JC, Hildebrand BA, Sun M, et al. A clinical frailty index in aging mice: comparisons with frailty index data in humans. J Gerontol Ser Biomed Sci Med Sci 2013;69:621–32.
103 Deepa SS, Bhaskaran S, Espinoza S, et al. A new mouse model of frailty: the Cu/Zn superoxide dismutase knockout mouse. Geroscience 2017;39:187–98.
104 Garatachea N, Pareja-Galeano H, Sanchis-Gomar F, et al. Exercise attenuates the major hallmarks of aging. Rejuvenation Res 2015;18:57–89.
105 Duggal NA, Niemiro G, Harridge SDR, et al. Can physical activity ameliorate immunosenescence and thereby reduce age-related multi-morbidity? Nat Rev Immunol 2019;19:563–72.
106 Quach A, Levine ME, Tanaka T, et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging 2017;9:419–46.
107 Justice JN, Ferrucci L, Newman AB, et al. A framework for selection of blood-based biomarkers for geroscience-guided clinical trials: report from the TAME Biomarkers Workgroup. GeroScience 2018;40:419–36.
108 Johnson SC, Kaeberlein M. Rapamycin in aging and disease: maximizing efficacy while minimizing side effects. Oncotarget 2016;7:44876–8.
109 Timmers S, Konings E, Bilet L, et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab 2011;14:612–22.
110 Madeo F, Carmona-Gutierrez D, Hofer SJ, et al. Caloric Restriction Mimetics against Age-Associated Disease: Targets, Mechanisms, and Therapeutic Potential. Cell Metab 2019;29:592–610.
111 Florea V, Bagno L, Rieger AC, et al. Attenuation of frailty in older adults with mesenchymal stem cells. Mech Ageing Dev 2019;181:47–58.
112 Morley JE. Senolytics: The Modern Snake Oil? J Nutr Health Aging 2019;23:490–3.