
J. Cummings1, K. Blennow2, K. Johnson3, M. Keeley4, R.J. Bateman5, J.L. Molinuevo6, J. Touchon7, P. Aisen8, B. Vellas9 and the EU/US/CTAD Task Force*
* EU/US/CTAD TASK FORCE: EU/US/CTAD TASK FORCE: Bjorn Aaris Gronning (Valby); Paul Aisen (San Diego); John Alam (Cambridge); Sandrine Andrieu (Toulouse), Randall Bateman (St. Louis); Monika Baudler (Basel); Joanne Bell (Wilmington); Kaj Blennow (Mölndal); Claudine Brisard (Blue Bell); Samantha Budd-Haeberlein (USA); Szofia Bullain (Basel) ; Marc Cantillon (Princeton) ; Maria Carrillo (Chicago); Gemma Clark (Princeton); Jeffrey Cummings (Las Vegas); Daniel Di Giusto (Basel); Rachelle Doody (Basel); Sanjay Dubé (Aliso Viejo); Michael Egan (North Wales); Howard Fillit (New York); Adam Fleisher (Philadelphia); Mark Forman (North Wales); Cecilia Gabriel-Gracia (Suresnes); Serge Gauthier (Montreal); Jeffrey Harris (South San Francisco); Suzanne Hendrix (Salt Lake City); Dave Henley (Titusville); David Hewitt (Blue Bell); Mads Hvenekilde (Basel); Takeshi Iwatsubo (Tokyo); Keith Johnson (Boston); Michael Keeley (South San Francisco); Gene Kinney (South San Francisco); Ricky Kurzman (Woodcliffe Lake); Valérie Legrand (Nanterre); Stefan Lind (Valby); Hong Liu-Seifert (Indianapolis); Simon Lovestone (Oxford); Johan Luthman (Woodcliffe); Annette Merdes (Munich); David Michelson (Cambridge); Mark Mintun (Philadelphia); José Luis Molinuevo (Barcelona); Susanne Ostrowitzki (South San Francisco); Anton Porsteinsson (Rochester); Martin Rabe (Woodcliffe Lake); Rema Raman (San Diego); Elena Ratti (Cambridge); Larisa Reyderman (Woodcliffe Lake); Gary Romano (Titusville); Ivana Rubino (Cambridge); Marwan Noel Sabbagh (Las Vegas); Stephen Salloway (Providence); Cristina Sampaio (Princeton); Rachel Schindler (USA); Peter Schüler (Langen); Dennis Selkoe (Boston); Eric Siemers (Indianapolis); John Sims (Indianapolis); Heather Snyder (Chicago); Georgina Spence (Galashiels); Bjorn Sperling (Valby); Reisa Sperling (Boston); Andrew Stephens (Berlin); Joyce Suhy (Newark); Gilles Tamagnan (New Haven); Edmond Teng (South San Francisco); Gary Tong (Valby); Jan Torleif Pedersen (Valby); Jacques Touchon (Montpellier); Bruno Vellas (Toulouse ); Vissia Viglietta (Cambridge) ; Christian Von Hehn (Cambridge); Philipp Von Rosenstiel (Cambridge) ; Michael Weiner (San Francisco); Kathleen Welsh-Bohmer (Durham); Iris Wiesel (Basel); Haichen Yang (North Wales); Wagner Zago (South San Francisco); Beyhan Zaim (Woodcliffe Lake); Henrik Zetterberg (Mölndal)
1. University of Nevada Las Vegas, School of Allied Health Sciences and Cleveland Clinic Lou Ruvo Center for Brain Health, Las Vegas, Nevada, USA; 2. Inst. of Neuroscience and Physiology, University of Gothenburg, Sahlgrenska University Hospital, Mölndal, Sweden; 3. Massachusetts General Hospital, Harvard Medical School, Boston MA, USA; 4. Genentech Research and Early Development, So. San Francisco, CA, USA; 5. Washington University School of Medicine, St. Louis, MO, USA; 6. BarcelonaBeta Brain Research Center Pasqual Maragall Foundation, Barcelona, Spain; 7. Montpellier University, and INSERM U1061, Montpellier, France; 8. Alzheimer’s Therapeutic Research Institute (ATRI), Keck School of Medicine, University of Southern California, San Diego, CA, USA; 9. Gerontopole, INSERM U1027, Alzheimer’s Disease Research and Clinical Center, Toulouse University Hospital, Toulouse, France
Corresponding Author: Jeffrey Cummings, University of Nevada Las Vegas, School of Allied Health Sciences and Cleveland Clinic Lou Ruvo Center for Brain Health, Las Vegas, Nevada, USA, cumminj@ccf.org
J Prev Alz Dis 2019;
Published online April 18, 2019, http://dx.doi.org/10.14283/jpad.2019.14
Abstract
Efforts to develop effective disease-modifying treatments for Alzheimer’s disease (AD) have mostly targeted the amyloid β (Aβ) protein; however, there has recently been increased interest in other targets including phosphorylated tau and other forms of tau. Aggregated tau appears to spread in a characteristic pattern throughout the brain and is thought to drive neurodegeneration. Both neuropathological and imaging studies indicate that tau first appears in the entorhinal cortex and then spreads to the neocortex. Anti-tau therapies currently in Phase 1 or 2 trials include passive and active immunotherapies designed to prevent aggregation, seeding, and spreading, as well as small molecules that modulate tau metabolism and function. EU/US/CTAD Task Force members support advancing the development of anti-tau therapies, which will require novel imaging agents and biomarkers, a deeper understanding of tau biology and the dynamic interaction of tau and Aβ protein, and development of multiple targets and candidate agents addressing the tauopathy of AD. Incorporating tau biomarkers in AD clinical trials will provide additional knowledge about the potential to treat AD by targeting tau.
Key words: Alzheimer’s disease, tau, tauopathy, therapeutics, biomarkers.
Introduction
No new drugs have been approved by the US Food and Drug Administration (FDA) for the treatment of Alzheimer’s disease (AD) since 2003 (1) despite the fact that an estimated 5.7 million Americans and 50 million people worldwide have AD today, and the prevalence is expected to grow to 152 million worldwide by 2050 (2, 3). AD clinical trials have failed at a very high rate: between 2002 and 2012, 99.6% of AD drugs tested failed to demonstrate clinical efficacy (1). Possible reasons for the high failure rate include targeting the wrong pathology or the wrong stage of disease (4, 5). Inappropriately designed trials and other methodological or unknown factors may have also contributed to treatment failures (6).
Despite the disappointments of the past 20 years, many experts in the Alzheimer’s community see reasons for optimism, including the emergence of novel drugs addressing a broader array of mechanisms than in the past (7). A recent report on the status of the AD drug development pipeline identified 112 agents: 26 in Phase 3 studies, 75 in Phase 2 studies, and 23 in Phase 1 studies (8). Moreover, whereas most of the negative studies in recent years targeted brain amyloidosis and amyloid β (Aβ), current studies are targeting a broader repertoire of mechanisms, including tau pathology. Of the 26 agents in Phase 3, only one targets tau, while 9 of the agents in Phase 2 (5 immunotherapies and 4 anti-aggregation agents) target tau (8).
Biology of tau and anti-tau therapeutics
The microtubule-associated protein tau, commonly referred to simply as tau, is found in a hyperphosphorylated form as insoluble, filamentous tangles and neuropil threads as well as dystrophic neurites in the AD brain (9). Along with plaques made up of aggregated Aβ protein, neurofibrillary tangles (NFTs) represent one of the hallmark pathologies of AD. Like Aβ, tau is found in several forms in the brain including monomers, oligomers, and fibrillary tangles (10). Tau pathology also plays a central role in other neurodegenerative diseases known collectively as tauopathies, including the primary tauopathies frontotemporal lobar degeneration with tau inclusions (FTLD-tau), Pick’s disease, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), argyrophilic grain disease (AGD), and chronic traumatic encephalopathy (CTE) (11).
Six isoforms of tau exist; it binds to several other proteins; and it undergoes many post-translational modifications, all of which contribute to its multiple functions in the brain. Tau protein plays important roles in cytoskeletal stability, cell signaling, synaptic plasticity, and neurogenesis (12, 13). In the AD brain, NFTs and neuropil threads composed of aggregated hyperphosphorylated tau are thought to be the primary drivers of neurodegeneration, although the mechanisms underlying the pathogenic process and the exact relationships of tau to Aβ remain unclear. Evidence also strongly suggests that tau propagates or spreads between cells (14) and that neuroinflammation triggered by microglial activation and astrogliosis contributes to tau-associated pathogenesis. Microglia may contribute to tau spreading (15). While postmortem and tau positron emission tomography (PET) studies indicate that tau spreading is associated with disease progression (16, 17), there are many unanswered questions regarding the rate of seeding or the effects of tau spreading on neuronal biology. If the spread of tau is driving clinical and cognitive changes, this would support intervening at the earliest stages of the tau-related disease process.
Neuropathological and imaging studies using PET suggest that tau aggregates are found in the entorhinal cortex and then the neocortex. If and how this drives neurodegeneration, what forms of tau are toxic, and the relationship of tau to amyloid in terms of toxicity remain unanswered questions. Tau pathology correlates much more closely to cognitive decline than does amyloid pathology (18, 19), and a recent study suggests that tau aggregation is linked to neurodegeneration and clinical manifestations of AD (20).
The complexity of tau biology provides many potential therapeutic targets to prevent tau production, aggregation, or spread at the level of transcription, phosphorylation, depolymerization, and transport. For example, preclinical studies indicate that antibodies against tau can prevent the trans-synaptic transmission of tau between neurons (21). A Phase 1 study of the humanized monoclonal antibody ABBV-8E12 showed acceptable safety and tolerability, which provided the basis for initiating a Phase 3 study in PSP patients to assess dose-related efficacy (22). The antibody is intended to prevent the trans-neuronal spread of the tau protein. Other monoclonal antibodies being assessed in early phase studies and targeting aspects of the tau protein include BIIB092, LY3303560, and RO7105705 (Table 1).
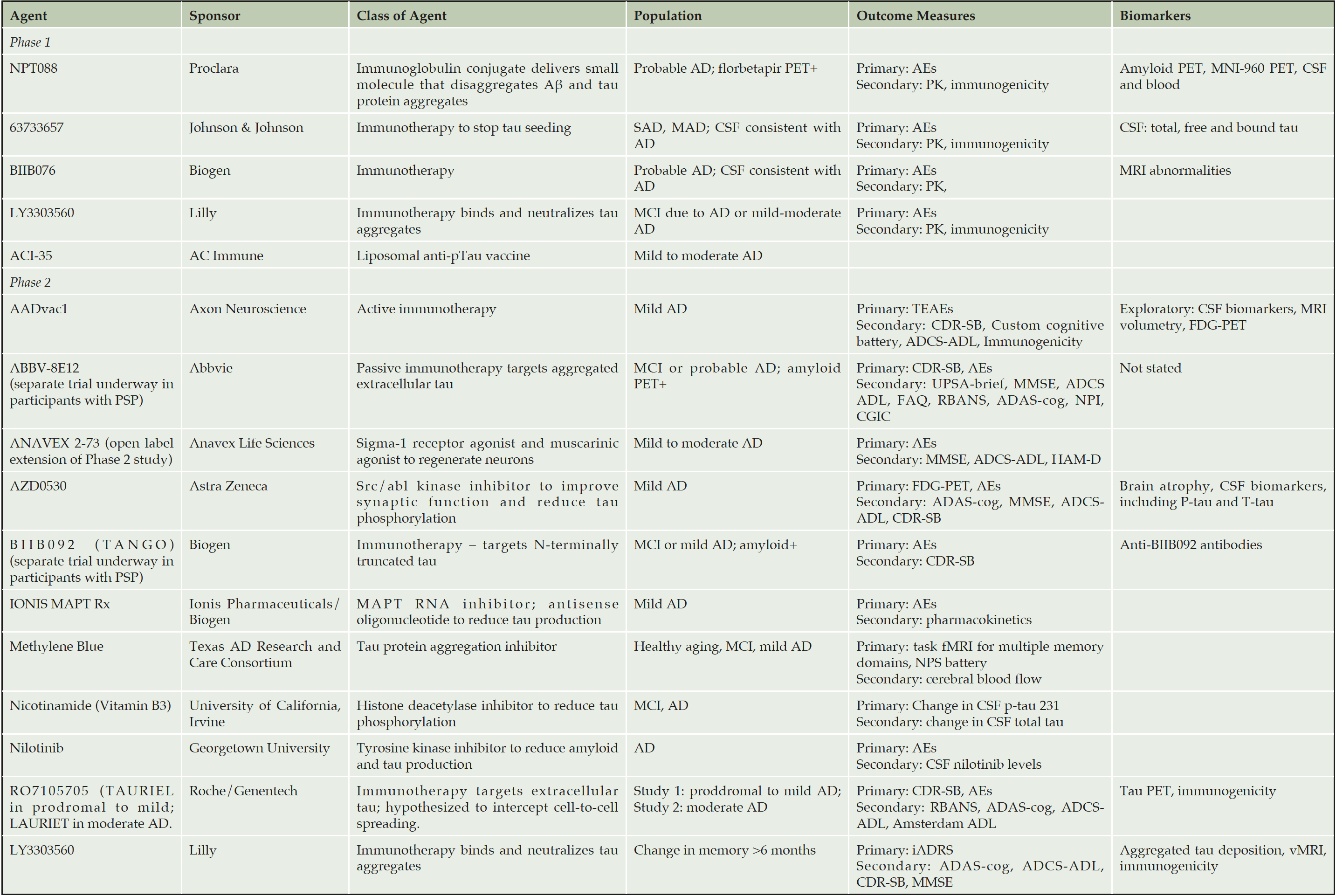
Table 1. Phase 1 and 2 clinical Trials targeting tau in AD populations
ADAS-cog = Alzheimer’s Disease Assessment Scale, cognition subscale, ADCS-ADL = Alzheimer’s Disease Cooperative Study – Activities of Daily Living Inventory, AEs= adverse events, CDR-SB = Clinical Dementia Rating Scale Sum of Boxes, CGIC = Clinical Global Impressions Scale, FAQ = functional activities questionnaire, fMRI = functional magnetic resonance imaging, HAM-D = Hamilton Psychiatric Rating Scale for Depression, iADRS=integrated AD rating scale, MMSE = Mini-mental state examination, NPI = neuropsychiatric inventory, NPS battery = neuropsychiatric symptoms battery, RBANS= Repeatable Battery for the Assessment of Neuropsychological Status, TEAEs = treatment-emergent adverse events. UPSA=University of California Performance Based Skills Assessment, brief version
Current status of anti-tau therapies in the AD treatment pipeline
One putative anti-tau agent, TRx0237 was studied in a Phase 3 trial and failed to show a difference between different doses. Studies in mouse models suggested that the agent functioned as an aggregation inhibitor and reduced the number of tau positive neurons (23); no target engagement biomarker was included in trial to determine if this was achieved in humans (24). Subgroup analyses suggest that some patients may have benefited from therapy and further studies of this compound are underway (24). Table 1 summarizes the anti-tau agents that are currently being tested in Phase 1 or Phase 2 clinical trials. These include both passive and active immunotherapies with monoclonal antibodies as well as drugs that affect the molecular structure of tau to modulate its function or prevent phosphorylation.
Other anti-tau drugs are also in development for AD including epigallocatechin-3 gallate (EGCG), a polyphenolic flavanoid extracted from green tea 25, and AC Immune’s tau morphomers, small molecules designed to inhibit aggregation and seeding and disaggregate already formed tau aggregates. Preclinical studies suggest that tau morphomers reduce pathological tau, improve cognition and function, and reduce microglia activation. Importantly, they are capable of crossing the blood-brain barrier.
Outcome measures and biomarkers
Tau PET imaging
Tau and Aβ aggregates in the brain have been investigated in several cohort studies, both neuropathologically at autopsy and in living people using PET (26-28). The overall picture emerging from these studies is that among cognitively normal individuals, about one-third have high amyloid, and among those with high amyloid about half also have high tau loads. A minority of cognitively normal individuals have sub-threshold levels of amyloid and high tau. The anatomic location of tau deposition may be important. These observations raise the possibility that quantifying progression of tau pathology may provide an early indicator of disease.
Johnson and colleagues have investigated the anatomical variability of amyloid and tau deposition in more than 400 individuals. These data indicate that distribution of tau in the rhinal cortex correlates with amyloid burden and that low amyloid individuals just starting to show elevations in tau are those most likely to be on the way to neocortical tauopathy. By the time tau levels have increased in the inferior temporal cortex, individuals may show significant impairments. These data support the hypothesis that amyloid is associated with tau spread.
Longitudinal data also provide support for these measures as useful for staging in order to establish a basis on which to measure change in serial imaging that could be useful in the clinical and clinical trial settings. Four stages were proposed:
Stage 0 – No signal exceeding background, consistent with Braak 0.
Stage 1 – Rhinal cortex signal emerging in a minority of low-amyloid clinically unimpaired individuals (allocortex, MTL) consistent with Braak I/II
Stage 2 – Inferior temporal signal emerging in the presence of high levels of fibrillar amyloid in clinically unimpaired individuals (corresponding to Braak stages III/VI)
Stage 3 – Additional neocortical binding in mild cognitive impairment (MCI) and AD patients (beyond inferior temporal; corresponding to Braak stages V/VI)
Figure 1 provides an example of images with high and low tau burden.
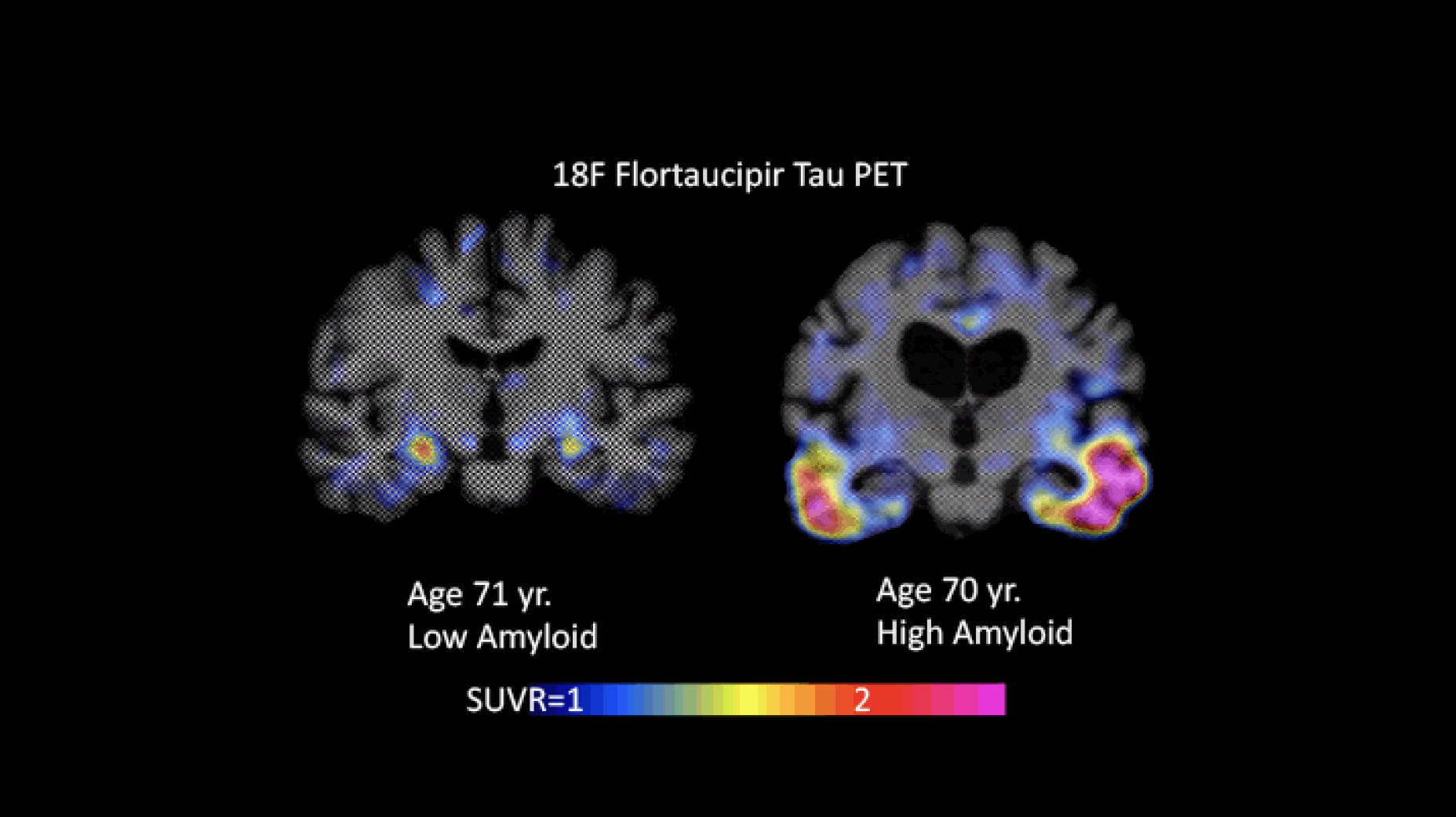
Figure 1. Flortaucipir images with low (Braak I/II) and high (Braak III/VI) levels of tau. The individual whose image is shown on the left had low amyloid levels; the one shown on the right had high amyloid levels (images courtesy of Keith Johnson)
CSF and blood biomarkers of tau
A systematic review and meta-analysis of cerebrospinal fluid (CSF) and blood biomarkers showed that CSF levels of total tau (T-tau), phosphorylated tau (p-tau), Aβ42, and neurofilament light (NfL), and plasma levels of T-tau were associated with AD and MCI due to AD but with quite pronounced, assay-dependent variation between studies, and no or only weak correlation with CSF T-tau levels (29-31). With regard to P-tau, a semi-sensitive assay for tau phosphorylated at threonine 181 (similar to the most employed CSF test) with electrochemiluminescence detection has been developed (32). Using this assay, plasma P-tau concentration was higher in AD dementia patients than controls. Plasma P-tau concentration was associated with both Aβ and tau PET and more AD-associated than the corresponding plasma T-tau test, which are promising results in need of replication. While conventional plasma measures of Aβ42 and Aβ40 by ELISA do not show a consistent change in clinically diagnosed AD cases as compared with cognitively unimpaired elderly (29), recent studies of blood Aβ using single molecule array (Simoa) or mass spectrometry have shown a relationship between blood levels of Aβ 40/42 ratios and the brain burden of Aβ (33-35). NfL indicates axonal damage and can also be measured in blood (36). Blood NfL shows particular promise as a biomarker of neurodegeneration in AD (37, 38) but high levels are also found in many other disorders characterized by neurodegeneration (39, 40). Given that NfL is a general neurodegeneration marker and not specifically involved in AD pathophysiology, it may give more unbiased information than tau biomarkers in clinical trials. Furthermore, the correlation between CSF and blood levels of NfL is very high (36), which is not the case for blood measures of tau (30). Synaptic proteins, including dendritic protein neurogranin and the pre-synaptic growth-associated protein 43 (GAP-43), show marked increases in CSF and are seemingly specific for AD (41, 42). Emerging CSF biomarkers including neuron-specific enolase (NSE), visinin-like protein 1 (VLP-1), heart fatty acid binding protein (HFPAP), and YKL-40 (a marker of glial activation) show moderate associations with AD (29, 43).
CSF tau comprises many different tau fragments that reflect processing of secreted tau, and some of these fragments may prove to be useful diagnostically (44) or provide information about tau kinetics in neurons (45). New assays are being developed to measure additional endogenous tau fragments that may correlate with tau pathology. For example, one of these tau fragments, tau368, results from cleavage of tau by asparagine endopeptidase (AEP) at position 368. The result of this is tau hyperphosphorylation, impaired microtubule assembly, and aggregation of truncated tau in neurofibrillary tangles (46). Inhibiting AEP may represent a novel therapeutic strategy for neurodegenerative disease (47). Tau368 can be measured in CSF and a first small study shows an association with longitudinal increase in tau PET tracer retention (48). Further, mass spectrometry studies show that CSF tau is specifically cleaved to a mid-domain fragment between amino acids 222-225 (45). Using an assay based on an end-specific tau x-224 monoclonal antibody, increased CSF levels were found in AD, while tau224 levels were low in other tauopathies (49). Exosomal tau has been evaluated as a biomarker but the studies have not been replicated and it is presently not possible to draw any conclusion on whether or not exosomal tau is a biomarker for AD.
The varying measures of tau report on different aspects of AD biology. In the amyloid, tau, neurodegeneration (ATN) Framework for AD diagnosis (50), tau PET and CSF p-tau are viewed as reporters of the presence and spread of tau pathology, whereas CSF t-tau, fluorodeoxyglucose PET, and MRI atrophy are seen as reporters of neurodegeneration. Recent evidence suggests that the soluble forms of tau are increased in production with greater amyloid plaque burden (45), while aggregated forms of tau appear at later stages of AD pathophysiology, closer to symptom onset. Tau markers — tau PET, p-tau, t-tau — measure different aspects of AD from this perspective.
For use in clinical trials of anti-tau agents, CSF biomarkers of amyloid and tau are needed to provide evidence of target engagement, enable enrichment of trials with appropriate participants, and show downstream effects of treatment (51). Lowering of CSF p-tau may suggest an effect on tau phosphorylation; however, more studies are needed to evaluate how CSF p-tau relates to brain pathology. Biomarker studies in recent clinical trials of the anti-amyloid antibodies bapineuzumab, gantenerumab, and BAN-2401 suggest that declines in CSF p-tau, t-tau, neurogranin and NfL indicate a downstream effect of Aβ immunotherapy on neurodegeneration, tau pathology, and synaptic degeneration (52-54).
Fully automated CSF immunoassays of AD biomarkers are now available, and in a study comparing fully automated CSF immunoassay with amyloid PET imaging, a multinational group of investigators found that the CSF tau/Aβ ratio was as accurate as amyloid PET in predicting clinical progression among patients with MCI (55) .
Challenges and unanswered questions
While the development of tau-targeted therapies is seen by many in the AD research community as one of the highest priority efforts, the complexity of tau protein processing gives rise to many challenges that have slowed development of tau-based therapies (56). Among the questions raised by the Task Force were these:
• What is known about the normal physiological function of tau, and are there potential negative/untoward consequences of reducing tau?
• What is the effect of tau suppression on spatiotemporal deposition of tau?
• What degree of tau lowering should be targeted to achieve an optimal therapeutic effect?
• What other factors may contribute to tau-based neurodegeneration (e.g., inflammation, aging, or vascular factors?)
• What is the relationship of amyloid-beta and tau?
• What is the relationship of soluble forms of tau and aggregated tau deposits?
• What is the role of microglia activation in the development of tau pathology?
• Since most tau is intracellular, will targeting it extracellularly be sufficient; or is there a window of time during which limiting extracellular tau would show a treatment benefit?
• What happens downstream when an antibody binds to tau? Is it sequestered or disposed of through cellular mechanisms or the glymphatic system 57 and does this result in downstream preservation of neurons?
• What are the best tau epitopes or tau fragments to target?
• Which tau fragments correlate best with AD-type neurodegeneration in CSF or in plasma?
• Which p-tau variants in CSF or blood correlate best with tau pathology in AD, or can differentiate AD from other tauopathies?
• Are there differential rates of change in tau deposition across the anatomy?
• What regions should tau PET target to demonstrate target engagement, and how should tau PET be developed for use in clinical trials to predict treatment response or measure treatment effect?
• What will be required to make tau PET useful clinically for diagnosis, prognosis, or prediction of treatment response?
• Do trials for anti-tau agents require similar structures as for Aβ-targeting agents even though the dynamics of the protein are different?
• What is the best population, taking into account the ATN stage, to target?
• Should anti-tau clinical trials focus on subpopulations and if so, which subpopulations?
• Would the best path forward for anti-tau agents be to test them in combination trials with Aβ-targeting agents or drugs that target other pathologies such as neuroinflammation?
• How can tau-PET be used to stage AD?
• What are the best tau-related outcomes for AD trials?
Conclusions
Anti-tau therapies are beginning to populate the AD drug development pipeline, mostly in Phase 1 and Phase 2 trials. However, anti-tau treatments have not yet shown evidence of a treatment effect in patients. The Task Force concluded that the development of anti-tau treatment will be determined by multiple trials and will require contributions from industry, academia, and advocacy groups.
The Task Force also called for incorporating CSF tau measures in all anti-tau trials. At a later date, tau PET may also be a viable option. For a biomarker to accurately assess target engagement and for pharmacodynamic studies, assays need to be designed specifically for the therapeutic antibody in addition to general tau-based assays. Such assays would enable exploration of whether a change in a specific tau species indicates that the therapeutic antibody binds tau in the brain parenchyma and if bound tau is secreted into the CSF.
Most Task Force members agreed that anti-tau trials are justified because AD symptoms are likely driven by the spread of tau and its degenerative effects, as well as by amyloid. However, most members also agreed that the specific tau-based mechanisms that will likely provide a treatment effect from anti-tau therapy are unclear and that significant observational and trial related studies will help better inform which tau targets will be most effective.
Acknowledgements: The authors thank Lisa J. Bain for assistance in the preparation of this manuscript.
Conflicts of interest: The Task Force was partially funded by registration fees from industrial participants. These corporations placed no restrictions on this work. Dr. Cummings is the Chief Scientific Officer of CNS Innovations. He acknowledges funding from the National Institute of General Medical Sciences (Grant: P20GM109025) and support from Keep Memory Alive; Consultation for Pharmaceutical Companies: Dr. Cummings has provided consultation to Acadia, Accera, Actinogen, AgeneBio, Alkahest, Allergan, Alzheon, Avanir, Axsome, Binomics, BiOasis Technologies, Biogen, Bracket, Denali, Diadem, EIP Pharma, Eisai, Genentech, Green Valley, Grifols, Hisun, Idorsia, Lundbeck, MedAvante, Merck, Otsuka, Pain Therapeutics, Probiodrug, Proclara, QR, Resverlogix, Roche, Samumed, Shinkei Therapeutics, Sunovion, Suven, Takeda, and United Neuroscience pharmaceutical and assessment companies. Consultation for Foundations: Dr. Cummings has provided consultation to Global Alzheimer Platform (GAP). Stock: Dr. Cummings owns stock in ADAMAS, BioAsis, Prana, MedAvante, Neurokos, and QR Pharma. Board member: None. Speaker/lecturer: None. Other: Dr. Cummings owns the copyright of the Neuropsychiatric Inventory (NPI). Dr. Cummings is the Chief Scientific Officer of CNS Innovations.Expert witness/legal consultation: None. NIH support: COBRE grant # P20GM109025; TRC-PAD # R01AG053798; DIAGNOSE CTE # U01NS093334.Research Support: None. Spousal ownership or significant financial interest in a relevant company: CNS Innovations. Dr. Johnson has consulted for Merck, Eli Lilly, Novartis, Biogen, Takeda, Roche, Eisai, Piramal, and GE. Dr. Keeley reports that he is an employee of Genentech. Dr. Bateman reports grants from BrightFocus Foundation, Pharma Consortium (Abbvie, AstraZeneca, Biogen, Eisai, Eli Lilly and Co., Hoffman La-Roche Inc., Janssen, Pfizer, Sanofi-Aventi), the Tau SILK/PET Consortium (Biogen/Abbvie/Lilly), Association for Frontotemporal Degeneration FTD Biomarkers Initiative, Anonymous Foundation, GHR Foundation, NIH, Alzheimer’s Association, Lilly, Rainwater Foundation Tau Consortium, and Cure Alzheimer’s Fund, grants, personal fees and non-financial support from Roche and Janssen, personal fees and non-financial support from Pfizer, Eisai, and Merck, and non-financial support from Avid Radiopharmaceuticals outside the submitted work. Washington University, Dr. Bateman, and David Holtzman have equity ownership interest in C2N Diagnostics and receive royalty income based on technology (stable isotope labeling kinetics and blood plasma assay) licensed by Washington University to C2N Diagnostics. RJB receives income from C2N Diagnostics for serving on the scientific advisory board. Washington University, with RJB as co-inventor, has submitted the US nonprovisional patent application “Methods for Measuring the Metabolism of CNS Derived Biomolecules In Vivo” and provisional patent application “Plasma Based Methods for Detecting CNS Amyloid Deposition”. Dr. Molinuevo reports personal fees from Alergan, from Oryzon, from Genentech, from Novartis, from Lundbeck, from Biogen, from Lilly, from Janssen, Green Valley, from MSD, from Eisai, from Alector and from Raman Health, outside the submitted work. Dr. Touchon has nothing to disclose. Dr. Aisen reports grants from Lilly, personal fees from Proclara, other from Lilly, other from Janssen, other from Eisai, grants from Janssen, grants from NIA, grants from FNIH, grants from Alzheimer’s Association, personal fees from Merck, personal fees from Roche, personal fees from Lundbeck, personal fees from Biogen, personal fees from ImmunoBrain Checkpoint, outside the submitted work. Dr. Vellas reports grants from Lilly, Merck, Roche, Lundbeck, Biogen, grants from Alzheimer’s Association, European Commission, personal fees from Lilly, Merck, Roche, Biogen, outside the submitted work.
References
1. Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther 2014;6:37.
2. Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimers Dement 2018;14:367-429.
3. Alzheimer’s Disease International. World Alzheimer’s Report 2018. The state of the art of dementia research: New frontiers. London2018.
4. Sperling RA, Jack CR, Jr., Aisen PS. Testing the right target and right drug at the right stage. Sci Transl Med 2011;3:111cm133.
5. Vellas B, Sampaio C, Bateman R, et al. EU./U.S. CTAD Task Force on Alzheimer’s Trial Populations. J Prev Alzheimers Dis 2014;1:110-116.
6. Ousset PJ, Cummings J, Delrieu J, et al. Is Alzheimer’s Disease Drug Development Broken? What Must Be Improved. J Prev Alzheimers Dis 2014;1:40-45.
7. Aisen PS. Continuing Progress in Alzheimer’s Disease Trials: Cause for Optimism. J Prev Alzheimers Dis 2017;4:211-212.
8. Cummings J, Lee G, Ritter A, Zhong K. Alzheimer’s disease drug development pipeline: 2018. Alzheimers Dement (N Y) 2018;4:195-214.
9. Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 2011;1:a006189.
10. Cardenas-Aguayo Mdel C, Gomez-Virgilio L, DeRosa S, Meraz-Rios MA. The role of tau oligomers in the onset of Alzheimer’s disease neuropathology. ACS Chem Neurosci 2014;5:1178-1191.
11. Holtzman DM, Carrillo MC, Hendrix JA, et al. Tau: From research to clinical development. Alzheimers Dement 2016;12:1033-1039.
12. Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol 2017;133:665-704.
13. Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron 2011;70:410-426.
14. Mudher A, Colin M, Dujardin S, et al. What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol Commun 2017;5:99.
15. Leyns CEG, Holtzman DM. Glial contributions to neurodegeneration in tauopathies. Mol Neurodegener 2017;12:50.
16. Braak H, Del Tredici K. Spreading of Tau Pathology in Sporadic Alzheimer’s Disease Along Cortico-cortical Top-Down Connections. Cereb Cortex 2018;28:3372-3384.
17. Schwarz AJ, Yu P, Miller BB, et al. Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 2016;139:1539-1550.
18. van Rossum IA, Vos SJ, Burns L, et al. Injury markers predict time to dementia in subjects with MCI and amyloid pathology. Neurology 2012;79:1809-1816.
19. Roe CM, Fagan AM, Grant EA, et al. Amyloid imaging and CSF biomarkers in predicting cognitive impairment up to 7.5 years later. Neurology 2013;80:1784-1791.
20. Ossenkoppele R, Schonhaut DR, Scholl M, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 2016;139:1551-1567.
21. Lee SH, Le Pichon CE, Adolfsson O, et al. Antibody-Mediated Targeting of Tau In Vivo Does Not Require Effector Function and Microglial Engagement. Cell Rep 2016;16:1690-1700.
22. West T, Hu Y, Verghese PB, et al. Preclinical and Clinical Development of ABBV-8E12, a Humanized Anti-Tau Antibody, for Treatment of Alzheimer’s Disease and Other Tauopathies. J Prev Alzheimers Dis 2017;4:236-241.
23. Melis V, Magbagbeolu M, Rickard JE, et al. Effects of oxidized and reduced forms of methylthioninium in two transgenic mouse tauopathy models. Behav Pharmacol 2015;26:353-368.
24. Gauthier S, Feldman HH, Schneider LS, et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 2016;388:2873-2884.
25. Gueroux M, Fleau C, Slozeck M, Laguerre M, Pianet I. Epigallocatechin 3-Gallate as an Inhibitor of Tau Phosphorylation and Aggregation: A Molecular and Structural Insight. J Prev Alzheimers Dis 2017;4:218-225.
26. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 2012;8:1-13.
27. Jack CR, Jr., Wiste HJ, Weigand SD, et al. Age-specific and sex-specific prevalence of cerebral beta-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50-95 years: a cross-sectional study. Lancet Neurol 2017;16:435-444.
28. Sepulcre J, Grothe MJ, Sabuncu M, et al. Hierarchical Organization of Tau and Amyloid Deposits in the Cerebral Cortex. JAMA Neurol 2017;74:813-820.
29. Olsson B, Lautner R, Andreasson U, et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol 2016;15:673-684.
30. Mattsson N, Zetterberg H, Janelidze S, et al. Plasma tau in Alzheimer disease. Neurology 2016;87:1827-1835.
31. Zetterberg H, Wilson D, Andreasson U, et al. Plasma tau levels in Alzheimer’s disease. Alzheimers Res Ther 2013;5:9.
32. Mielke MM, Hagen CE, Xu J, et al. Plasma phospho-tau181 increases with Alzheimer’s disease clinical severity and is associated with tau- and amyloid-positron emission tomography. Alzheimers Dement 2018;14:989-997.
33. Nakamura A, Kaneko N, Villemagne VL, et al. High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature 2018;554:249-254.
34. Ovod V, Ramsey KN, Mawuenyega KG, et al. Amyloid beta concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement 2017;13:841-849.
35. Janelidze S, Stomrud E, Palmqvist S, et al. Plasma beta-amyloid in Alzheimer’s disease and vascular disease. Sci Rep 2016;6:26801.
36. Gisslen M, Price RW, Andreasson U, et al. Plasma Concentration of the Neurofilament Light Protein (NFL) is a Biomarker of CNS Injury in HIV Infection: A Cross-Sectional Study. EBioMedicine 2016;3:135-140.
37. Lewczuk P, Ermann N, Andreasson U, et al. Plasma neurofilament light as a potential biomarker of neurodegeneration in Alzheimer’s disease. Alzheimers Res Ther 2018;10:71.
38. Zetterberg H, Skillback T, Mattsson N, et al. Association of Cerebrospinal Fluid Neurofilament Light Concentration With Alzheimer Disease Progression. JAMA Neurol 2016;73:60-67.
39. Bacioglu M, Maia LF, Preische O, et al. Neurofilament Light Chain in Blood and CSF as Marker of Disease Progression in Mouse Models and in Neurodegenerative Diseases. Neuron 2016;91:56-66.
40. Hansson O, Janelidze S, Hall S, et al. Blood-based NfL: A biomarker for differential diagnosis of parkinsonian disorder. Neurology 2017;88:930-937.
41. Portelius E, Olsson B, Hoglund K, et al. Cerebrospinal fluid neurogranin concentration in neurodegeneration: relation to clinical phenotypes and neuropathology. Acta Neuropathol 2018;136:363-376.
42. Sandelius A, Portelius E, Kallen A, et al. Elevated CSF GAP-43 is Alzheimer’s disease specific and associated with tau and amyloid pathology. Alzheimers Dement 2019;15:55-64.
43. Molinuevo JL, Ayton S, Batrla R, et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol 2018;136:821-853.
44. Meredith JE, Jr., Sankaranarayanan S, Guss V, et al. Characterization of novel CSF Tau and ptau biomarkers for Alzheimer’s disease. PLoS One 2013;8:e76523.
45. Sato C, Barthelemy NR, Mawuenyega KG, et al. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 2018;97:1284-1298 e1287.
46. Zhang Z, Song M, Liu X, et al. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat Med 2014;20:1254-1262.
47. Zhang Z, Kang SS, Liu X, et al. Asparagine endopeptidase cleaves alpha-synuclein and mediates pathologic activities in Parkinson’s disease. Nat Struct Mol Biol 2017;24:632-642.
48. Leuzy A, Cicognola C, Chiotis K, et al. Longitudinal tau and metabolic PET imaging in relation to novel CSF tau measures in Alzheimer’s disease. Eur J Nucl Med Mol Imaging 2019.
49. Cicognola C, Brinkmalm G, Wahlgren J, et al. Novel tau fragments in cerebrospinal fluid: relation to tangle pathology and cognitive decline in Alzheimer’s disease. Acta Neuropathol 2019;137:279-296.
50. Jack CR, Jr., Bennett DA, Blennow K, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018;14:535-562.
51. Blennow K, Zetterberg H. Amyloid and Tau Biomarkers in CSF. J Prev Alzheimers Dis 2015;2:46-50.
52. Blennow K, Zetterberg H, Rinne JO, et al. Effect of immunotherapy with bapineuzumab on cerebrospinal fluid biomarker levels in patients with mild to moderate Alzheimer disease. Arch Neurol 2012;69:1002-1010.
53. Ostrowitzki S, Lasser RA, Dorflinger E, et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther 2017;9:95.
54. Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 2014;370:322-333.
55. Hansson O, Seibyl J, Stomrud E, et al. CSF biomarkers of Alzheimer’s disease concord with amyloid-beta PET and predict clinical progression: A study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement 2018;14:1470-1481.
56. Khanna MR, Kovalevich J, Lee VM, Trojanowski JQ, Brunden KR. Therapeutic strategies for the treatment of tauopathies: Hopes and challenges. Alzheimers Dement 2016;12:1051-1065.
57. Rasmussen MK, Mestre H, Nedergaard M. The glymphatic pathway in neurological disorders. Lancet Neurol 2018;17:1016-1024.