
T. West1, Y. Hu1, P.B. Verghese1, R.J. Bateman2, J.B. Braunstein1, I. Fogelman1, K. Budur3, H. Florian3, N. Mendonca4, D.M. Holtzman2
1. C2N Diagnostics LLC, Saint Louis, MO, USA; 2. Washington University, St. Louis, MO, USA; 3. AbbVie Inc., North Chicago, IL, USA; 4. AbbVie Deutschland GmbH & Co. KG, Ludwigshafen, Germany
Corresponding Author: Tim West, PhD, C2N Diagnostics, 20 S Sarah St, Saint Louis, MO 63108, Email: twest@c2ndiagnostics.com
J Prev Alz Dis 2017;4(4):236-241
Published online September 27, 2017, http://dx.doi.org/10.14283/jpad.2017.36
Abstract
Tau neurofibrillary tangles are found in the brains of patients suffering from Alzheimer’s disease and other tauopathies. The progressive spreading of tau pathology from one brain region to the next is believed to be caused by extracellular transsynaptic transmission of misfolded tau between neurons. Preclinical studies have shown that antibodies against tau can prevent this transfer of misfolded tau between cells. Thus, antibodies against tau have the potential to stop or slow the progression of tau pathology observed in human tauopathies. To test this hypothesis, a humanized anti-tau antibody (ABBV-8E12) was developed and a phase 1 clinical trial of this antibody has been completed. The double-blind, placebo-controlled phase 1 study tested single doses of ABBV-8E12 ranging from 2.5 to 50 mg/kg in 30 patients with progressive supranuclear palsy (PSP). ABBV-8E12 was found to have an acceptable safety profile with no clinically concerning trends in the number or severity of adverse events between the placebo and dosed groups. Pharmacokinetic modelling showed that the antibody has a plasma half-life and cerebrospinal fluid:plasma ratio consistent with other humanized antibodies, and there were no signs of immunogenicity against ABBV-8E12. Based on the acceptable safety and tolerability profile of single doses of ABBV-8E12, AbbVie is currently enrolling patients into two phase 2 clinical trials to assess efficacy and safety of multiple doses of ABBV-8E12 in patients with early Alzheimer’s disease or PSP.
Key words: Tau, immunotherapy, Alzheimer’s disease, tauopathy, therapeutic.
Rationale
Tauopathies refer to a set of neurodegenerative disorders characterized by the pathological aggregation of microtubule-associated protein tau (MAPT) in neurons and glial cells in the human brain. The most common tauopathy is Alzheimer’s disease (AD), estimated to affect more than 5 million Americans and 46 million people worldwide. Less common tauopathies include progressive supranuclear palsy (PSP), cortical basal degeneration (CBD), Pick’s disease and frontotemporal dementia with parkinsonism linked to chromosome 17, with the total number of tauopathies being greater than 20 (1). In some of these tauopathies (such as PSP, CBD, and Pick’s), aggregation of tau in the brain is the predominant form of brain pathology, while for other diseases (such as AD and Niemann Pick type C) other brain pathologies are involved in the disease alongside tau.
The human tau gene contains 16 exons and alternative spicing of exons 2, 3 and 10 results in six different tau protein isoform (2). Based on the number of inserts near the amino-terminal and the carboxy-terminal repeat domains, the isoforms are referred to as 0N3R, 1N3R, 2N3R, 0N4R, 1N4R and 2N4R. Under normal conditions, tau is predominantly localized within neurons and more specifically axons, although non-neuronal cells can have trace amounts (3). Tau was originally identified as a microtubule-associated protein that functions to promote assembly of microtubule protein subunit tubulin into microtubules and stabilize their structure (4). More recently, novel functions of tau have been discovered, such as iron transport, neurogenesis, synaptic plasticity and neuronal DNA protection (5).
Data from biochemical and animal studies of the tau protein suggest a working model that can serve as a basic framework for studying the pathophysiological functions of tau as well as for developing tau therapeutics. Tauopathy pathogenesis includes molecular events such as hyperphosphorylation and aggregation of tau. Hyperphosphorylation of tau can reduce its binding to microtubules, and thereby cause microtubule disassembly and axonal transport impairment and eventually synaptic dysfunction (6, 7). Hyperphosphorylation, as well as some other modifications of tau (such as truncation and O-glcNAcylation) also promote tau aggregation (5).
Aggregated tau can assume the form of either soluble oligomers, insoluble paired helical filaments (PHFs), or insoluble straight filaments. PHFs manifest as neurofibrillary tangles (NFT) in the brain and are one of the major histopathological hallmarks of tauopathies. While NFTs were originally assumed to be toxic and the main cause of neurodegeneration, growing evidence suggests that NFT are neither necessary nor sufficient to cause neurodegeneration (8–10).
Recent evidence suggests that of the various aggregated tau species, tau oligomers can be toxic to neurons (11–15) and that release of tau oligomers from neurons can lead to transmission of tau pathology between cells. The tau oligomers are taken up by synaptically connected neurons, causing the normal tau in the recipient neuron to aggregate – thus resulting in spreading of tau pathology from one neuron to the next through neuronal connectivity. Calvaguera et al. found that intracerebral injection of brain extract from mice with filamentous tau pathology induces the formation and spreading of aggregates made of hyperphosphorylated tau in mice expressing human wild-type tau (16). Subsequently, more studies have reported similar findings in vivo, confirming this “prion-like” property of pathological tau species isolated from human tissue or transgenic mice (17–21). This mode of transcellular propagation suggests that extracellular transfer of tau between cells may be a susceptible target for antibody-mediated therapies. In support of this hypothesis, tau immunotherapy has emerged as a promising therapeutic strategy for tauopathies and this approach has been shown to reduce tau pathology and improve behavioral deficits in animal models (22–27).
Preclinical data
The laboratory of Dr. Holtzman at Washington University School of Medicine generated a library of anti-human tau antibodies to test the hypothesis that propagation of aggregated tau between cells can be prevented by using anti-tau antibodies and to further assess the mechanisms of action of such antibodies (28). The library originated from mice immunized with full-length human tau protein (2N4R) and antibodies recognizing various epitopes of human tau were identified. Using an in vitro cell based assay, the anti-tau antibodies were found to specifically and dose dependently block uptake of misfolded tau from brain lysates into neuronal cells (19, 28).
To assess the in vivo activity of the antibodies, three of the anti-tau antibodies and one control antibody were administered intracerebroventricularly (ICV) into P301S tau transgenic mice. The P301S transgenic mouse carries a mutated human tau gene that causes early onset frontotemporal dementia in humans. These mice develop brain tau pathology as evidenced by presence of NFTs and phosphorylated tau as well as behavioral deficits consistent with human tau pathology (29). Antibodies were infused continuously starting just after the time when tau pathology starts to develop in these mice (6 months of age). At nine months, associative learning was assessed and the brains of the animals were assessed for a variety of histological and biochemical measures. Of the three antibodies tested in vivo, one antibody (HJ8.5) demonstrated a consistent effect on all of the outcome measures as compared to the control antibody. This antibody showed a reduction in phosphorylated tau by both biochemical and histopathological measures, and an improvement in associative learning (28).
In addition to evaluating central delivery of the antibody, the biological effects of administering HJ8.5 peripherally were assessed in a separate study. Here, six-month old P301S mice received weekly intraperitoneal (IP) doses of PBS, or 10 or 50 mg/kg of HJ8.5 for three months (30). After three months of treatment, there was a significant reduction of tau pathology in the hippocampus in both the 10 mg/kg and 50 mg/kg dose group compared with control (Figure 1A and B). This reduction in tau pathology was matched by a reduction in the formic acid insoluble tau measured in the cortex of P310S mice in the 50 mg/kg dose group (Figure 1C). Peripheral HJ8.5 administration also improved sensorimotor function in these mice as measured by inverted screen and ledge tests. Importantly, peripheral antibody administration significantly attenuated brain volume loss observed in P301S mice over the three-month timeframe. This finding is significant since brain atrophy in human tauopathies associates strongly with tau accumulation in the brain regions affected, and thus it appears that the reduction in tau pathology by the antibody treatment results in protection against brain atrophy.
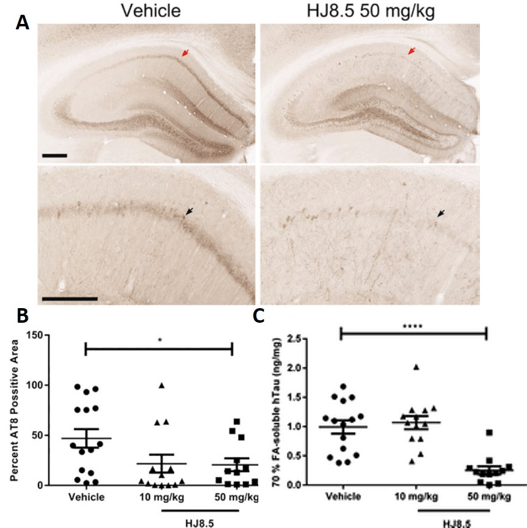
Figure 1. Anti-tau antibody decreased phospho-tau staining in the hippocampal CA1 cell layer
(A) Representative coronal sections of biotinylated AT8 antibody staining of phosphorylated tau in the hippocampal CA1 cellular region of 9-month-old P301S mice treated for three months with vehicle and HJ8.5 at 50 mg/kg. The lower images are higher power views of the CA1 region in the uppers panels. Red arrows indicate the area magnified in the lower image. Black arrows indicate the hippocampal CA1 cell layer. (B) Quantification of biotinylated AT8 antibody staining of abnormally phosphorylated tau revealed a significant decrease in AT8 staining in mice treated with HJ8.5 at 50 mg/kg in the hippocampal CA1 cellular layer compared to vehicle-treated mice (P = 0.035). Values represent mean ± SEM. (C) Levels of formic acid soluble tau determined by ELISA. HJ8.5 treatment at 50 mg/kg significantly decreased insoluble human tau (P < 0.0001) compared to vehicle-treated mice. Values represent mean ± SEM. ****P < 0.0001. Figure is modified from (30) with permission.
At the end of the peripheral dosing study, the concentration of tau in the plasma of P310S mice was measured using a tau ELISA. The concentration of tau in plasma was significantly increased in P310S animals that had been injected with HJ8.5, and the increase was higher in the animals that received the higher dose of HJ8.5 (30). Further studies indicated that the antibody dependent increase of plasma tau resulted primarily from CNS derived tau and that plasma tau concentrations appeared to reflect soluble, extracellular tau in the brain (31).
Recent studies utilized adenoassociated virus (AAV) to express single chain fragment variable domains (scFv) derived from HJ8.5 directly in the brain of P301S transgenic mice (32). This treatment also decreased tau pathology, demonstrating that the Fc domain of the antibody is not required for a therapeutic effect.
In summary, these preclinical studies show that anti-tau antibodies have the potential to engage tau present in the brain’s extracellular space as well as in plasma. Administration of the anti-tau antibody HJ8.5 in these animal models via central (ICV) and peripheral (IP) routes markedly reduced tau pathology, neuronal loss, and brain atrophy, resulting in cognitive and sensorimotor preservation compared to control treated mice.
Clinical data
Based on the promising results from the transgenic mouse model studies, a variety of humanized anti-tau antibodies were generated to test the hypothesis that anti-tau antibodies can provide therapeutic benefits in human tauopathies. Following requisite cell line development and toxicology testing, clinical trials with the lead compound (C2N-8E12, now known as ABBV-8E12) were initiated during the summer of 2015. A phase 1 clinical trial (NCT02494024) was designed to test the safety and tolerability of a single dose of ABBV-8E12 in patients with PSP. Key inclusion and exclusion criteria for this study are listed in Table 1. Subjects were randomized in blocks of four in a double-blind manner to receive a single intravenous dose of ABBV-8E12 or placebo in a three to one ratio (drug:placebo). Using a continual reassessment method that pre-specified algorithms for dose escalation, each block of subjects was assigned to one of the five dose cohorts (2.5, 7.5, 15, 25, and 50 mg/kg) tested in the study. Dose escalation was implemented only after available safety data from lower doses had been reviewed by the data safety monitoring committee (DSMC). Safety was monitored for 84 days post-dosing and included AEs, laboratory analyses, MRI assessments, ECG evaluations, physical/neurological examinations, vital signs, mental health assessments and a brain MRI at two weeks after dosing.
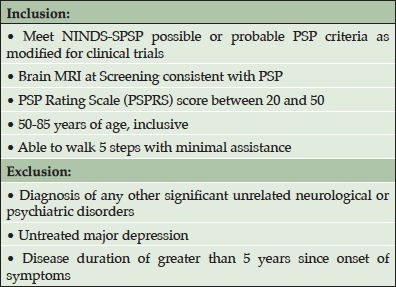
Table 1. Key inclusion and exclusion criteria for the phase 1 clinical trial (NCT02494024)
A total of 38 subjects were screened for the phase 1 trial, with 30 subjects enrolling. Of the enrolled subjects, 7 were assigned to the placebo arm and 23 were assigned to one of the 5 dose arms (Table 2).The safety profile at all doses supported dose escalation to the maximum dose (50 mg/kg), which was administered to 10 subjects. Twenty-seven subjects completed the 84-day follow-up and one subject withdrew from the study due to an adverse event (AE). AEs occurred in 21 of the 30 (70%) study participants. Table 3 provides an overview of the AEs observed in the study. The majority of the AEs were rated by the blinded investigators as mild or moderate in severity. Only two AEs were rated as severe – one case of headache and one case of agitation. Treatment-relatedness of the AEs was also rated by the investigators, with the majority of the AEs being rated as unrelated to treatment.
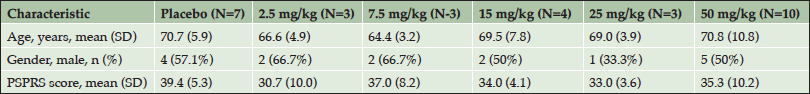
Table 2. Patient demographics and disease characteristics at screening
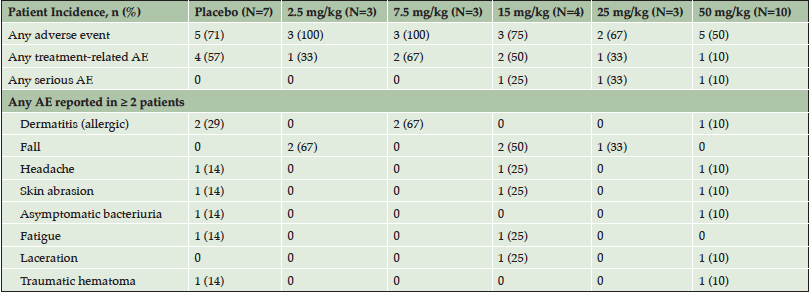
Table 3. Adverse event summary
Events are listed by MedDRA preferred term. Number of patients experiencing an event is shown in the table with the percent incidence in parentheses.
There were no clinically concerning trends observed in the number or severity of AEs between the placebo and ABBV-8E12 dose groups. Nearly half (44%) of the observed AEs resolved within the first two days of onset and 77% of AEs resolved within the first two weeks. At the day 14 MRI, no clinically significant radiographic abnormal findings were observed. Three serious adverse events (SAEs) were reported during the study. One subject, in the 15 mg/kg dose group, with a history of experiencing several falls at baseline, had subdural hematoma. One subject, in the 25 mg/kg dose group, with a history of anxiety and agitation around stressful events including medical procedures, reported an increase in anxiety, agitation and perseverative behaviors. One subject, in the 50 mg/kg dose group, with a history of hypertension had hypertensive cerebrovascular disease. These events did not indicate an emerging safety issue. Thus, ABBV-8E12, when administered in single doses of up to 50 mg/kg, appears to have an acceptable safety and tolerability profile.
While assessment of single-dose safety and tolerability were the primary objectives of this phase 1 study, both plasma and CSF samples were acquired for assessment of pharmacokinetics (PK) of ABBV-8E12 in plasma and penetration of drug into the brain. Figure 2 shows the average PK profiles for ABBV-8E12 in plasma for each dose cohort. Across the dose range of 2.5 mg/kg to 50 mg/kg IV, the ABBV-8E12 area under the curve (AUC) increased in a dose-proportional manner. The harmonic mean plasma half-life ranged from approximately 27 days to 37 days. This plasma half-life is consistent with what has been reported for other monoclonal antibodies.
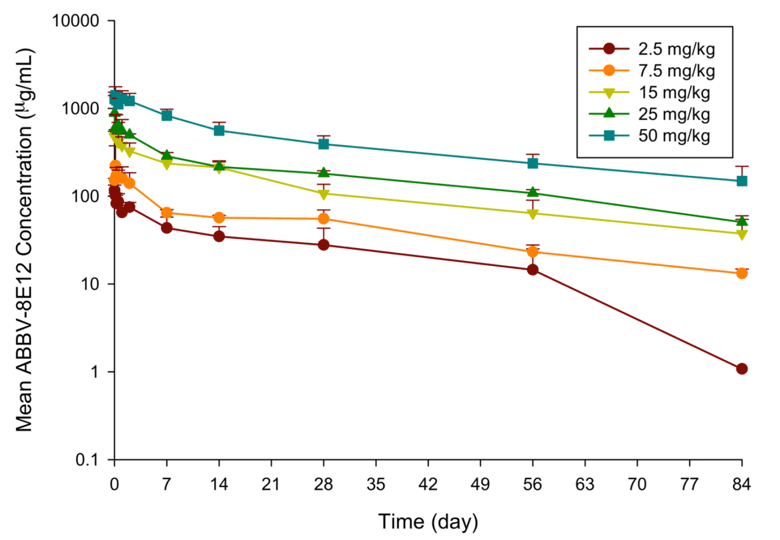
Figure 2. Pharmacokinetic profiles of ABBV-8E12 in plasma. Plasma samples were collected for up to 84 days post dosing and drug concentrations measured using a validated PK assay
Data shown as mean with error bars representing the standard deviation.
Cerebrospinal fluid (CSF) was sampled at screening and 14 days after drug administration. Measurement of ABBV-8E12 in CSF on day 14 revealed that CSF ABBV-8E12 concentrations increased with dose. Comparison of CSF concentrations to plasma concentrations of ABBV-8E12 on the same day showed that the CSF/plasma ratio ranged from 0.181% to 0.385%, consistent with what has been observed in other studies of monoclonal antibodies. Plasma samples collected out to day 84 were also assayed for the presence of anti-ABBV-8E12 antibodies using a validated anti-drug antibody assay. No anti-drug antibodies were detected in the post-dose plasma samples analyzed.
Future development plan/discussion
Based on the acceptable safety and tolerability profile of single doses up to 50 mg/kg in PSP patients, AbbVie is currently enrolling patients into two phase 2 clinical trials that assess the efficacy and safety of multiple doses of ABBV-8E12 in patients with early AD or PSP. In these studies, patients will be dosed for 96 weeks (early AD) or 52 weeks (PSP). The first cohort of 48 and 30 subjects each, in AD and PSP studies respectively, will undergo intensive safety monitoring.
The phase 2 AD study is a randomized, double-blind, placebo-controlled trial designed to evaluate the efficacy and safety of ABBV-8E12 in patients with early AD. For the purposes of this study, early AD is defined as a score of 22 or higher on the mini-mental state examination, a global score of 0.5 on clinical dementia rating, a score of 85 or lower on the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS)-delayed memory index, and a positive amyloid positron emission tomography (PET) scan.
The phase 2 AD study consists of a screening period of up to 8 weeks, a 96 week double-blind treatment period and a follow-up period of approximately 20 weeks following the last study drug administration. Approximately 400 subjects will be enrolled to meet the study objectives. Eligible subjects are between 55 to 85 years of age and have early AD in the absence of concurrent diseases that could confound safety or efficacy evaluations. Study participants are allowed to use concomitant medications to treat symptoms related to AD, if they are on a stable dose for at least 12 weeks prior to randomization. Upon completion of screening and baseline procedures, eligible subjects are randomized to one of the 3 ABBV-8E12 dose arms (low, medium and high dose) or placebo. Doses are administered every 4 weeks via IV infusion.
The primary efficacy measure for the AD study is the Clinical Dementia Rating scale – Sum of Boxes (CDR-SB). Secondary efficacy and exploratory outcomes include a variety of clinical measures, biologic markers, and neuroimaging measures. Safety will be monitored by adverse event reports, physical examination, laboratory tests, and imaging.
The phase 2 PSP study is a multiple dose, multicenter, multinational, randomized, double-blind, placebo-controlled trial designed to evaluate the efficacy and safety of ABBV-8E12 in patients with PSP. Eligible subjects are randomized to one of the two ABBV-8E12 dose arms (low and high dose) or placebo and ABBV-8E12 is administered every 4 weeks via IV infusion for a total of a 52-week treatment period. The study starts with a screening period of up to 8 weeks and ends with a follow-up period of approximately 20 weeks following the last study drug administration. Approximately 180 subjects will be enrolled to meet the study objectives. Eligible subjects are 40 years of age or older and meet PSP clinical criteria in the absence of concurrent diseases that could confound safety or efficacy evaluations. Study participants are required to have PSP symptoms for less than 5 years and to be able to walk 5 steps with minimal assistance.
The primary efficacy measure of this PSP phase 2 study is the PSP Rating Scale (PSPRS) (33). Secondary efficacy measures include a variety of biologic markers and clinical and neuroimaging outcome measures. Safety is assessed by adverse event reports, physical examination, laboratory tests and imaging.
Funding: This work was funded by C2N Diagnostics and AbbVie Inc., and supported by a grant from the Alzheimer’s Association (PCTR-15-330406) made possible by Part the Cloud™.
Acknowledgements: The team at C2N Diagnostics and AbbVie Inc. would like to sincerely thank the clinical investigators and the patients in the phase 1 clinical study for all their hard work and dedication to this clinical trial.
Conflict of interest: TW, IHH, PBV, and IF are full time employees and/or advisors of C2N Diagnostics, receiving stock and/or stock options. RJB is a co-founder of C2N Diagnostics. RJB and Washington University in St. Louis have equity ownership interest in C2N Diagnostics and may receive royalty income based on technology licensed by Washington University to C2N Diagnostics. RJB receives lab research funding from the Tau SILK Consortium (AbbVie, Biogen, and Eli Lilly and Co.), Eli Lilly and Co, Hoffman La-Roche, Janssen, Avid Radiopharmaceuticals, and the DIAN Pharma Consortium (Abbvie, Amgen, AstraZeneca, Biogen, Eisai, Eli Lilly and Co, Hoffman La-Roche, Janssen, Pfizer, and Sanofi). RJB has received honoraria from Janssen and Pfizer as a speaker and from Merck and Pfizer as an Advisory Board member. In addition, RJB receives income from C2N Diagnostics for serving on the Scientific Advisory Board. JBB is a co-founder and employee of C2N Diagnostics, receiving stock and/or stock options. KB, HF, and NM are employees of AbbVie, receiving stock and/or stock options. DMH co-founded and is on the scientific advisory board of C2N Diagnostics. DMH is an inventor on a submitted patent “Antibodies to Tau”, PCT/US2013/049333, that is licensed by Washington University to C2N Diagnostics. This patent was subsequently licensed to AbbVie. DMH receives research grants from the C2N Diagnostics, AbbVie, Eli Lilly, and Denali. DMH consults for Genentech, AbbVie, Eli Lilly, Proclara Biosciences, Glaxosmithkline, and Denali.
References
1. Williams DR. Tauopathies: classification and clinical update on neurodegenerative diseases associated with microtubule-associated protein tau. Intern Med J 2006;36:652–60. doi:10.1111/j.1445-5994.2006.01153.x.
2. Andreadis A, Broderick JA, Kosik KS. Relative exon affinities and suboptimal splice site signals lead to non-equivalence of two cassette exons. Nucleic Acids Res 1995;23:3585–93.
3. Schoenfeld TA, Obar RA. Diverse distribution and function of fibrous microtubule-associated proteins in the nervous system. Int Rev Cytol 1994;151:67–137. doi:10.1016/S0074-7696(08)62631-5.
4. Drubin DG, Kirschner MW. Tau protein function in living cells. J Cell Biol 1986;103:2739–46. doi:10.1083/jcb.103.6.2739.
5. Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci 2015;17:22–35. doi:10.1038/nrn.2015.1.
6. Cho JH, Johnson GVW. Glycogen synthase kinase 3β phosphorylates tau at both primed and unprimed sites: Differential impact on microtubule binding. J Biol Chem 2003;278:187–93. doi:10.1074/jbc.M206236200.
7. Schneider A, Biernat J, Von Bergen M, Mandelkow E, Mandelkow EM. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 1999;38:3549–58. doi:10.1021/bi981874p.
8. Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005;309:476–81. doi:10.1126/science.1113694.
9. Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, et al. Tau-Induced Defects in Synaptic Plasticity, Learning, and Memory Are Reversible in Transgenic Mice after Switching Off the Toxic Tau Mutant. J Neurosci 2011;31:2511–25. doi:10.1523/JNEUROSCI.5245-10.2011.
10. Van Der Jeugd A, Hochgräfe K, Ahmed T, Decker JM, Sydow A, Hofmann A, et al. Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol 2012;123:787–805. doi:10.1007/s00401-012-0987-3.
11. Lasagna-Reeves C a, Castillo-Carranza DL, Sengupta U, Sarmiento J, Troncoso J, Jackson GR, et al. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J 2012;26:1946–59. doi:10.1096/fj.11-199851.
12. Maeda S, Sahara N, Saito Y, Murayama M, Yoshiike Y, Kim H, et al. Granular tau oligomers as intermediates of tau filaments. Biochemistry 2007;46:3856–61. doi:10.1021/bi061359o.
13. Tian H, Davidowitz E, Lopez P, Emadi S, Moe J, Sierks M. Trimeric tau is toxic to human neuronal cells at low nanomolar concentrations. Int J Cell Biol 2013. doi:10.1155/2013/260787.
14. Flach K, Hilbrich I, Schiffmann A, Gärtner U, Krüger M, Leonhardt M, et al. Tau oligomers impair artificial membrane integrity and cellular viability. J Biol Chem 2012;287:43223–33. doi:10.1074/jbc.M112.396176.
15. Tepper K, Biernat J, Kumar S, Wegmann S, Timm T, Hübschmann S, et al. Oligomer formation of tau protein hyperphosphorylated in cells. J Biol Chem 2014;289:34389–407. doi:10.1074/jbc.M114.611368.
16. Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 2009;11:909–13. doi:10.1038/ncb1901.
17. Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A 2013;110:9535–40. doi:10.1073/pnas.1301175110.
18. Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, et al. Distinct Tau Prion Strains Propagate in Cells and Mice and Define Different Tauopathies. Neuron 2014:1–18. doi:10.1016/j.neuron.2014.04.047.
19. Holmes BB, Furman JL, Mahan TE, Yamasaki TR, Mirbaha H, Eades WC, et al. Proteopathic tau seeding predicts tauopathy in vivo. Proc Natl Acad Sci U S A 2014;111:E4376-85. doi:10.1073/pnas.1411649111.
20. Guo JL, Narasimhan S, Changolkar L, He Z, Stieber A, Zhang B, et al. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J Exp Med 2016;213:2635 LP-2654. doi:10.1084/jem.20160833.
21. Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, et al. Trans-synaptic spread of tau pathology in vivo. PLoS One 2012;7. doi:10.1371/journal.pone.0031302.
22. Sigurdsson EM. Immunotherapy targeting pathological tau protein in Alzheimer’s disease and related tauopathies. J Alzheimer’s Dis 2008;15:157–168.
23. Sigurdsson EM. Tau-focused immunotherapy for Alzheimer’s disease and related tauopathies. Curr Alzheimer Res 2009;6:446–50.
24. Asuni A a, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci 2007;27:9115–29. doi:10.1523/JNEUROSCI.2361-07.2007.
25. Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp Neurol 2010;224:472–85. doi:10.1016/j.expneurol.2010.05.010.
26. Troquier L, Caillierez R, Burnouf S, Fernandez-Gomez FJ, Grosjean M-E, Zommer N, et al. Targeting phospho-Ser422 by active Tau Immunotherapy in the THYTau22 mouse model: a suitable therapeutic approach. Curr Alzheimer Res 2012;9:397–405. doi:10.2174/156720512800492503.
27. Bi M, Ittner A, Ke YD, Götz J, Ittner LM. Tau-targeted immunization impedes progression of neurofibrillary histopathology in aged P301L tau transgenic mice. PLoS One 2011;6. doi:10.1371/journal.pone.0026860.
28. Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, et al. Anti-Tau Antibodies that Block Tau Aggregate Seeding In Vitro Markedly Decrease Pathology and Improve Cognition In Vivo. Neuron 2013;80:402–14. doi:10.1016/j.neuron.2013.07.046.
29. Yoshiyama Y, Higuchi M, Zhang B, Huang S-M, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007;53:337–51. doi:10.1016/j.neuron.2007.01.010.
30. Yanamandra K, Jiang H, Mahan TE, Maloney SE, Wozniak DF, Diamond MI, et al. Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann Clin Transl Neurol 2015;2:278–88. doi:10.1002/acn3.176.
31. Yanamandra K, Patel TK, Jiang H, Schindler S, Ulrich JD, Boxer AL, et al. Anti-tau antibody administration increases plasma tau in transgenic mice and patients with tauopathy. Sci Transl Med 2017;9. doi:10.1126/scitranslmed.aal2029.
32. Ising C, Gallardo G, Leyns CEG, Wong CH, Stewart F, Koscal LJ, et al. AAV-mediated expression of anti-tau scFvs decreases tau accumulation in a mouse model of tauopathy. J Exp Med 2017;214:1227–38. doi:10.1084/jem.20162125.
33. Golbe LI, Ohman-Strickland P a. A clinical rating scale for progressive supranuclear palsy. Brain 2007;130:1552–65. doi:10.1093/brain/awm032.