
J. Alam, K. Blackburn, D. Patrick
EIP Pharma LLC, 11 Channing Street, Cambridge MA 02138, USA
Corresponding Author: John Alam, MD, EIP Pharma, LLC, 11 Channing Street, Cambridge MA 02138, phone: 617-945-0794, jalam@eippharma.com
J Prev Alz Dis 2017;4(4):273-278
Published online September 27, 2017, http://dx.doi.org/10.14283/jpad.2017.41
Abstract
Neflamapimod (previously code named VX-745) is a clinical phase 2b-ready highly specific inhibitor of the intra-cellular enzyme p38 mitogen activated protein kinase alpha (“p38α”) that is being developed as a disease-modifying drug for Alzheimer’s disease (AD) that acts via targeting synaptic dysfunction. Neflamapimod was discovered through a proprietary structure-based drug discovery platform at Vertex Pharmaceuticals, and developed previously by Vertex through to phase 2a in rheumatoid arthritis. EIP Pharma licensed the compound in 2014 for development and commercialization as a treatment of central nervous system (CNS) disorders. Neflamapimod is the most advanced in the clinic drug that targets specific molecular mechanisms within neurons that leads to synaptic dysfunction, the pathogenic process that is now considered to be a major driver of the development of memory deficits and disease progression in the early stages of AD. Based on the scientific rationale of targeting synaptic dysfunction and the preclinical data, neflamapimod has the potential to both reverse memory deficits and slow disease progression. Phase 2a clinical data in patients with early-stage AD (MMSE 20-28, biomarker positive) provides evidence that the preclinical science may be translatable to human Alzheimer’s, as 6- to 12-weeks of neflamapimod treatment led to significant improvement in episodic memory, the best clinical measure of synaptic dysfunction in AD. A phase 2b six-month placebo-controlled 150-patient clinical study is anticipated to start by end of 2017. This study is designed to definitively demonstrate that neflamapimod reverses memory deficits, and also to provide preliminary evidence that the drug slows disease progression.
Key words: Neflamapimod, p38α, synaptic dysfunction, episodic memory.
Abbreviations: AD: Alzheimer’s disease; APP: amyloid precursor protein; CDR-SB: Clinical Dementia Rating Scale-Sum of Boxes; CSF: Cerebrospinal fluid; ES: Effect Size; FCSRT: Free and Cued Selective Reminding Test; HVLT-R: Hopkins Verbal Learning Test – Revised; IC50: 50% inhibitory concentration; MMSE: Mini-Mental State Examination; PS1: presenilin 1; WMS: Wechsler Memory Scale.
Introduction
There is currently no therapy available for AD that reverses and/or slows disease progression. Until recently, the primary objective of experimental approaches to address disease progression in AD has been directed at slowing the rate of neuronal loss (“neurodegeneration”). As neurodegeneration is irreversible, an inherent limitation of such approaches is that they will be limited to slowing disease progression, rather than also reversing disease progression. In the past five years, dysfunction and loss of synapses has emerged as an additional therapeutic objective to addressing AD progression, particularly in the early stages of symptomatic disease prior to the onset of significant neurodegeneration. Specifically, it appears the synapse is the convergence point for amyloid beta, tau, and inflammation, with synaptic dysfunction and synapse loss potentially being the fundamental pathogenic event within the neuron that leads to the defining characteristic of AD, memory deficits (1). The importance of synaptic dysfunction was underscored in a recent review that stated “Several lines of evidence point to synaptic dysfunction as a cause of AD” (2).
Importantly, for AD disease progression, targeting synaptic dysfunction has potential advantages over targeting neurodegeneration. In particular, in animal models synaptic dysfunction and loss is reversible. Further, reversal of synaptic dysfunction in the early stages of clinical disease leads to both improvement in function (i.e., reversal of disease progression) that is measurable within weeks of treatment initiation and arrests the neurodegenerative process (3, 4). Thus, therapeutic interventions that target synaptic dysfunction have the potential to both reverse and slow disease progression in the early stages of AD, when neurodegeneration is not the primary driver of disease progression.
In parallel with the developments on the importance of synaptic dysfunction, as discussed in the scientific rational section, the understanding that the alpha isoform of the protein kinase p38 MAP kinase (p38α) as a critical driver of synaptic dysfunction in pre-clinical models has emerged (5-9). This article summarizes the scientific rationale for targeting p38α to reverse synaptic dysfunction in AD, and the non-clinical and clinical data with an investigational drug, neflamapimod (previously code-named VX-745) (10), that specifically inhibits p38α kinase activity. Collectively, the scientific rationale, nonclinical data and clinical results, indicate that neflamapimod has the potential to reverse AD-related synaptic dysfunction; in particular, the early clinical results strongly suggest that the mechanistic rationale is translating in the clinic, with improvement in episodic memory function with 6- 12- weeks of treatment. Confirmation of the ability of neflamapimod to reverse AD-related synaptic dysfunction awaits the results of a six-month placebo-controlled phase 2b study that is due to commence at the end of 2017.
Scientific Rationale
In the brain, p38α regulates inflammation through its expression in microglia and astrocytes. Under stress and disease, p38α is also expressed in neurons, where its expression is considered to be a critical contributor in the toxicity of amyloid-beta, inflammation, and tau to synapses (11-17). Consistent with that science, the functional deficits are fully reversed with only 2 to 3 weeks of treatment with p38α selective small molecule kinase inhibitors in three distinct animal models (APP/PS1, aged rats and hTau mice) in which cognitive deficits are induced by amyloid-beta, inflammation, or tau, respectively (18-20). Further, genetic reduction of neuronal p38α in Amyloid-Precursor-Protein (APP) overexpressing transgenic mice improves synaptic transmission and plasticity (i.e. prevents synaptic dysfunction), reduces memory loss, and reduces amyloid pathology (21). Moreover, genetically knocking down p38α in neurons protected mice from developing age-related hippocampal dysfunction and decline in neurogenesis (22).
From a mechanism standpoint, emerging evidence indicates the p38α mediates synaptic dysfunction and amyloid beta generation by disrupting proteostasis, i.e. the normal physiologic homeostatic regulation of protein turnover, within the neuron; while maintenance of proteostasis is critical for finely tuned synaptic function (23-25). More specifically, p38α has been demonstrated to impair autophagy-mediated protein degradation and endolysosomal function (25-28), both of which impair protein receptor turnover and protein synthesis.
Non-clinical results with neflamapimod
Potent, selective inhibitor of p38α kinase
Neflamapimod is an ATP competitive inhibitor of p38α kinase. In vitro, neflamapimod potently inhibits p38α, with a 9 nanomolar IC50 (50% inhibitory concentration) (10). When evaluated against a panel of 442 kinases by an independent academic laboratory VX-745 was shown to potently bind only p38α and p38β, the latter approximately 25-fold less potently (29). Based on these and other data, neflamapimod has been described as “the most selective” p38α inhibitor (30).
Potent cellular activity
In an in vitro human cell system, neflamapimod has a <10 nanomolar estimated potency (IC50) for reversing Alzheimer’s Precursor Protein (APP) induced endolysosomal dysfunction (28), the physiologic defect within neurons that is increasingly considered the major cause of synaptic dysfunction in AD (25). In the same experiments, the cellular potency for inhibition of p38 activity was less than 10 nanomolar concentration.
Reverses Synaptic Function In Vivo
To confirm the results with genetic knockout and other compounds on the role of p38α in the development of synaptic dysfunction, neflamapimod was tested in the aged rat model of age-related cognitive decline. The cognitive deficit in aged rats has been shown to result from synaptic dysfunction primarily due to inflammation (13), but also due to age-related increases in amyloid beta in the brain (31). Thus, aged rats provided an ideal platform to test whether neflamapimod would reverse synaptic dysfunction due to inflammation and amyloid-beta. The published results (19) showed that 17 days of treatment with neflamapimod fully reversed the learning deficits in the Morris-water-maze-test in 20- to 22-month old rats, with the performance of aged rats treated with neflamapimod at the optimal dose being significantly better than vehicle (placebo)- treated aged rats (p=0.007 for latency, p=0.01 for distance) and being similar to that of young rats. These data combined with dose response data in prior animal and clinical studies, were then utilized to identify doses for the phase 2a clinical studies in early AD.
Neflamapimod has also been studied in an induced-stroke model in rats: transient ischemia of sufficient duration was induced such that significant neurologic disability developed and did not substantially reverse during follow-up without therapy. These rats were then treated with vehicle (controls) or neflamapimod. Six weeks of neflamapimod treatment, starting at 48-hours after stroke, led to substantial improvement on multiple parameters of neurologic function compared to vehicle controls (p<0.001 for each of global neurologic scores, motor- and sensory-specific tests; Alam et al, manuscript submitted) (32). As recovery after stroke is dependent on neuronal and synaptic plasticity (33), these results further confirm that neflamapimod is active in reversing impaired synaptic plasticity in animals.
Toxicology
As described in detail in the neflamapimod investigator brochure, a full chronic toxicology program has been completed in rodents (rats) and non-rodents (dogs). In the rodent species, in the six-month toxicology study, no human relevant findings were evident at dose levels that provided plasma drug exposures approximately ten-fold higher than those achieved in the AD clinical trials; in shorter-term studies, the primary target organ was the liver, with findings commencing at plasma drug exposures twenty-fold higher than the AD clinical trial exposures. In the non-rodent species, in 9- and 12-month toxicology studies, dose dependent findings were evident beginning at plasma drug exposures approximately ten-fold higher than in AD clinical trials; with minimal to equivocal findings at that dose level in the liver, bone marrow and CNS.
Neflamapimod, p38α, synaptic dysfunction, episodic memory
Clinical results with neflamapimod
Prior Clinical Development Experience
Phase 1 trials and a phase 2a clinical trial in patients with rheumatoid arthritis (RA) were conducted by Vertex. Approximately 150 health volunteers or patients with RA received neflamapimod for up to 3 months at 250 mg twice daily, which is five times the therapeutic dose for AD. Anti-inflammatory effects and statistically significant effects on American College of Rheumatology response rates were demonstrated in the RA phase 2a clinical study (neflamapimod investigator brochure).
Early AD Phase 2a Clinical Trials Design
EIP Pharma initiated two phase 2a trials in patients in the early stages of Alzheimer’s disease in May 2015: Study 302, a 12-week treatment study conducted at VU Medical Center in Amsterdam, Netherlands; and Study 303, a 6-week treatment study conducted at the contract clinical pharmacology unit in Los Angeles. Both studies enrolled patients with Early Alzheimer’s disease, defined as patients with either Mild Cognitive Impairment (MCI) with biomarker evidence of Alzheimer’s (i.e. “MCI due to AD”) or very mild Alzheimer’s dementia. Oral doses of 40 mg and 125 mg twice daily in capsules were utilized in the studies. At baseline, MMSE ranged from 20 to 28, with a median of 24 in Study 302 and 25 in Study 303. The studies were completed in September 2016 and the main results have recently been reported at scientific conferences (CTAD, 2016; AD-PD, 2017; AAIC, 2017). The main findings are provided in Table 1.

Table 1. Neflamapimod Phase 2a Clinical Trials Findings
Pharmacokinetics (s), Pharmacodynamics (PD) and Safety Results in Phase 2a
The major PK, PD safety findings in the phase 2a clinical studies are as follows:
• Neflamapimod was confirmed to be robustly blood-brain-barrier (BBB) penetrant in humans, with a CSF to unbound plasma drug concentration ratio of ~1.2.
• Mechanistic target engagement in the brain was demonstrated for neflamapimod, as evidenced by (1) reductions in brain amyloid plaque load at the 40 mg dose level, assessed in Study 302 by quantitative dynamic amyloid PET scanning; and (2) plasma-drug concentration dependent reduction in CSF inflammatory markers (IL-8 and TNFα) in Study 303.
• Neflamapimod at a dose level up to 125 mg twice daily for 12 weeks was every well tolerated in patients with Alzheimer’s disease, with no safety signals identified. There was only one early treatment discontinuation (of 25 patients treated); which was attributed to side effects of lumbar puncture, rather than drug treatment. The most common treatment emergent adverse events were diarrhea and somnolence; 4 events each (16% incidence each), and all reported as mild.
As neuronal p38α has been shown to play a “critical role” in amyloid beta generation and plaque production (26,27), the effect on brain amyloid plaque load is likely due to inhibition of p38α within the neuron leading to decreased amyloid plaque production. At the higher dose level of 125 mg BID, this effect was not seen, potentially due to anti-inflammatory effect resulting from inhibition of p38 in microglia leading to reduction of amyloid plaque clearance, offsetting the effect on amyloid plaque production.
Cognitive (Episodic Memory) Results in Phase 2a Clinical Studies
From a clinical standpoint, the most important finding in the phase 2a studies was the result with the episodic memory tests given that the disruption of hippocampal-dependent episodic memory and learning is a critical event in the development and symptomatology of Alzheimer’s disease (34, 35). In clinical trials, this process is evaluated by providing specific verbal or visual information to a test subject and they are asked to either immediately provide the information back (“immediate recall”) or after a lag of 20 to 30 minutes (“delayed recall”). Importantly, none of the symptomatic therapies have demonstrated positive effects on episodic memory, perhaps because they do not impact underlying synaptic dysfunction.
Within the two phase 2a studies there were highly statistically significant within-subject improvements in tests of episodic memory and learning, as assessed by Wechsler Memory Scale (WMS) immediate and delayed recall composite measures in Study 302, and Hopkins Verbal Learning Test – Revised (HVLT-R) in Study 303. Analyses of the data also indicated the magnitude of the effects on these tests were clinically significant as the Effect Sizes (ES) of the treatment effects in the two studies were between 0.6 and 0.9, i.e. medium to large clinically significant treatment effects. Moreover, in clinical trials of other agents in comparable early-stage AD patients, in placebo-treated patients over 12 weeks on the WMS and HVLT-R demonstrate generally slight worsening; and when improvements have been seen, they are limited to an ES of less than 0.2 (36-39).
To further understand the impact of neflamapimod on episodic memory, an analysis of the relationship between plasma drug (neflamapimod) concentrations and outcome on WMS immediate and delayed recall tests in the 12-week study was conducted. Using a standard linear pharmacokinetics-pharmacodynamics (PK-PD) regression model, a highly significant positive correlation was established between plasma drug levels and level of improvement at Day 84 in combined WMS-immediate and delayed recall. Though full confirmation requires a placebo-controlled study the combination of the ES analysis (i.e. magnitude of treatment effect) and the PK-PD analysis strongly argue that the improvement seen in episodic memory in phase 2a was primarily due to neflamapimod treatment, and not due to chance or practice effects.
As the 125 mg dose level showed only modestly greater effect on episodic memory function and clearly less of an effect on amyloid plaques, 40 mg twice daily has been identified as the optimal dose for the treatment of Alzheimer’s disease.
Planned Phase 2b Clinical Study
The planned Phase 2b clinical study will be a six-month treatment randomized double-blind placebo-controlled proof-of-concept study in patients with early Alzheimer’s disease that would be designed to definitively demonstrate that neflamapimod reverses memory deficits, and in doing so, would be the first drug to show such an effect.
The main inclusion criteria, primary and secondary endpoints are shown in Table 2. Patients will be randomized to either neflamapimod 40 mg or placebo capsules twice daily with food for 24 weeks, with a sample size of 75 patients per treatment arm providing 90% power to detect effect size (ES) of .53 and 80% power to detect ES of .46. The sample size is substantially less than would be required if the treatment effect were to only slow decline in memory function; as it assumes an improvement of function with neflamapimod treatment, which compared to a drug that only slows decline increases the difference between it and the placebo group. The sample size is constructed so that as long as there is progression in the placebo group, any amount of improvement in the neflamapimod group will read out as a positive result with statistical significance; in every published study identified over six months there is deterioration in episodic memory in well defined early-stage AD patients.
After efficacy, the most common reason for failure in late-stage drug development is safety. In the case of neflamapimod, the risk of failure for safety reason should be less than other AD drugs at this stage of development because (1) a full chronic toxicology program has already been completed and there are robust safety margins, and (2) neflamapimod has already been evaluated at a substantially higher dose level, 250 mg twice daily, in the clinic for up to 3 months. In human clinical studies the dose-limiting toxicity of neflamapimod was liver enzyme elevations (transaminase only without bilirubin increases) that were seen in 10-15% of patients at a dose of 250 mg twice a day or higher. Liver enzyme elevations are a known class effect of p38 inhibitors, but are also known to be dose-dependent and seen only when drug levels exceed that required to inhibit cytokine production in the liver. At the 40 mg dose level, there is no to minimal systemic pharmacological activity outside the brain and consistent with that there were no effects on liver enzyme levels.
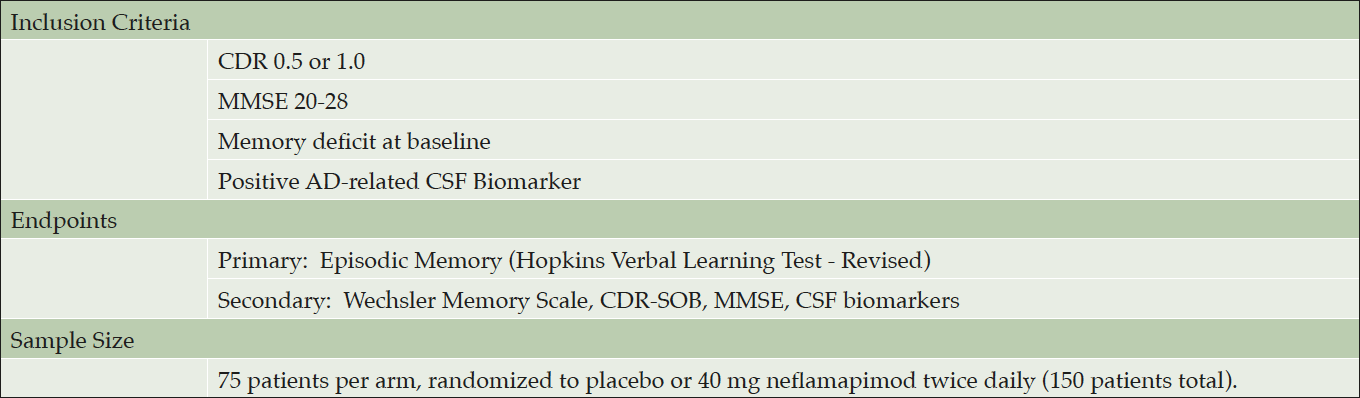
Table 2. Design of Planned Phase 2b Clinical Study
Patents
EIP Pharma has made a number of discoveries related to neflamapimod, that are reflected in four issued US patents on the administration of neflamapimod that are owned by the company: (a) #9,427,438, issued August 2016, to treat patients suffering from Alzheimer’s disease, (b) #8,697,627, issued April 2014, to lower brain amyloid plaque load (also issue of in EU and Japan); (c) #9,427,439, issued August 2016, to promote recovery of function in patients who have suffered acute neurologic injury, including that resulting from acute ischemic stroke; and (d) #9,579,322, issued February 2017, to improve cognition by administering sufficient drug to inhibit cytokine signaling, but not cytokine production. The terms of the above patents run to between 2031 and 2036. Additional patents have been filed in the US and internationally.
Conclusions
Neflamapimod through inhibiting p38α kinase activity has demonstrated potential to address synaptic dysfunction in AD. As synaptic dysfunction is considered to be the major driver of the development of memory deficits and disease progression in the early stages of AD, neflamapimod by targeting synaptic dysfunction has the potential to both reverse memory deficits and slow disease progression. In clinical studies to date, neflamapimod treatment in patients with early-stage AD led to potentially clinically meaningful and statistically significant improvement in episodic memory function, the best clinical measure of synaptic dysfunction in AD. Preparations are underway for a phase 2b placebo-controlled six-month treatment duration 150-patient clinical study that is designed to confirm the preliminary clinical findings by definitively demonstrating neflamapimod reverses memory deficits. This study could also provide preliminary evidence that the drug may slow disease progression.
Disclosures: The authors are affiliated with EIP Pharma LLC, the company that is developing neflamapimod as a disease-modifying therapy for AD.
Ethical standards: The manuscript conforms to the declaration and statements in the Publication Ethics section of the JPAD website. In addition, the referred to animal toxicology and clinical studies in the manuscript were conducted in accordance with global guidelines regarding Good Laboratory Practice (GLP) and Good Clinical Practice (GCP), respectively. For non-regulated animal studies, national guidelines were followed and relevant institutional animal use and care committee approval was obtained.
References
1. Spires-Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron. 2014; 82:756-771.
2. Nistico R, Pignatelli M, Piccinin S, Mercuri NB, Collingridge G. Targeting synaptic dysfunction in Alzheimer’s disease therapy. Mol Neurobiol. 2012; 46:572-587.
3. Freeman OJ, Mallucci GR. The UPR and synaptic dysfunction in neurodegeneration. Brain Res. 2016; 1648:530-537.
4. Smith HL, Mallucci GR. The unfolded protein response: mechanisms and therapy of neurodegeneration. Brain. 2016; 139:2113-2121.
5. Yasuda S, Sugiura H, Tanaka H, Takigami S, Yamagata K. p38 MAP kinase inhibitors as potential therapeutic drugs for neural diseases. Cent Nerv Syst Agents Med Chem. 2011; 11:45-59.
6. Correa SA, Eales KL. The Role of p38 MAPK and Its Substrates in Neuronal Plasticity and Neurodegenerative Disease. J Signal Transduct. 2012; 2012:649079.
7. Sanderson TM, Hogg EL, Collingridge GL, Correa SA. Hippocampal metabotropic glutamate receptor long-term depression in health and disease: focus on mitogen-activated protein kinase pathways. J Neurochem. 2016;139 Suppl 2:200-214.
8. Lauretti E, Li JG, Di Meco A, Pratico D. Glucose deficit triggers tau pathology and synaptic dysfunction in a tauopathy mouse model. Transl Psychiatry. 2017; 7:e1020.
9. Lee JK, Kim NJ. Recent advances in the inhibition of p38 MAPK as a potential strategy for the treatment of Alzheimer’s disease. Molecules 2017; 22:E1287.
10. Duffy JP, Harrington EM, Salituro FG, et al. The Discovery of VX-745: A Novel and Selective p38alpha Kinase Inhibitor. ACS Med Chem Lett. 2011; 2:758-763.
11. Li Y, Liu L, Barger SW, Griffin WS. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci. 2003; 23:1605-1611.
12. Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011; 31:6627-6638.
13. Lynch MA. Age-related neuroinflammatory changes negatively impact on neuronal function. Front Aging Neurosci. 2010; 1:6.
14. Watterson DM, Grum-Tokars VL, Roy SM, et al. Development of Novel In Vivo Chemical Probes to Address CNS Protein Kinase Involvement in Synaptic Dysfunction. PLoS One. 2013; 8:e66226.
15. Birnbaum JH, Bali J, Rajendran L, Nitsch RM, Tackenberg C. Calcium flux-independent NMDA receptor activity is required for Abeta oligomer-induced synaptic loss. Cell Death Dis. 2015; 6:e1791.
16. Koppensteiner P, Trinchese F, Fa M, et al. Time-dependent reversal of synaptic plasticity induced by physiological concentrations of oligomeric Abeta42: an early index of Alzheimer’s disease. Sci Rep. 2016;6:32553.
17. Prieto GA, Snigdha S, Baglietto-Vargas D, et al. Synapse-specific IL-1 receptor subunit reconfiguration augments vulnerability to IL-1beta in the aged hippocampus. Proc Natl Acad Sci U S A. 2015; 112:E5078-5087.
18. Roy SM, Grum-Tokars VL, Schavocky JP, et al. Targeting human central nervous system protein kinases: An isoform selective p38alphaMAPK inhibitor that attenuates disease progression in Alzheimer’s disease mouse models. ACS Chem Neurosci. 2015; 6:666-680.
19. Alam JJ. Selective Brain-Targeted Antagonism of p38 MAPKalpha Reduces Hippocampal IL-1beta Levels and Improves Morris Water Maze Performance in Aged Rats. J Alzheimers Dis. 2015; 48:219-227.
20. Maphis N, Jiang S, Xu G, et al. Selective suppression of the alpha isoform of p38 MAPK rescues late-stage tau pathology. Alzheimers Res Ther. 2016; 8:54.
21. Colie S, Sarroca S, Palenzuela R, et al. Neuronal p38alpha mediates synaptic and cognitive dysfunction in an Alzheimer’s mouse model by controlling beta-amyloid production. Sci Rep. 2017;7:45306.
22. Cortez I, Bulavin DV, Wu P, et al. Aged dominant negative p38alpha MAPK mice are resistant to age-dependent decline in adult-neurogenesis and context discrimination fear conditioning. Behav Brain Res. 2017; 322:212-222.
23. Scheper W, Hoozemans JJ. A new PERKspective on neurodegeneration. Sci Transl Med. 2013; 5:206fs237.
24. Nixon RA. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: inseparable partners in a multifactorial disease. Faseb j. 2017; 31:2729-2743.
25. Lachén-Montes M, González-Morales A, Zelaya MV, Pérez-Valderrama E, Ausín K, Ferrer I et al. Olfactory bulb neuroproteomics reveals a chronological perturbation of survival routes and a disruption of prohibit complex during Alzheimer’s disease progression. Sci Rep. 2017; 7:9115.
26. Schnoder L, Hao W, Qin Y, et al. Deficiency of Neuronal p38alpha MAPK Attenuates Amyloid Pathology in Alzheimer Disease Mouse and Cell Models through Facilitating Lysosomal Degradation of BACE1. J Biol Chem. 2016; 291:2067-2079.
27. Alam J, Scheper W. Targeting neuronal MAPK14/p38alpha activity to modulate autophagy in the Alzheimer disease brain. Autophagy. 2016; 12:2516-2520.
28. Alam J YJ, Nixon RA. Antagonism of p38 MAPK alpha (p38α) reverses APP-induced endosomal abnormalities and lysosomal dysfunction in Down Syndrome fibroblasts. Paper presented at: AAIC2017; London, UK.
29. Davis MI, Hunt JP, Herrgard S, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011; 29:1046-1051.
30. Uitdehaag JC, Verkaar F, Alwan H, de Man J, Buijsman RC, Zaman GJ. A guide to picking the most selective kinase inhibitor tool compounds for pharmacological validation of drug targets. Br J Pharmacol. 2012; 166:858-876.
31. Church RM, Miller MC, Freestone D, et al. Amyloid-beta accumulation, neurogenesis, behavior, and the age of rats. Behav Neurosci. 2014; 128:523-536.
32. Alam J, Krakovsky M, Lammensdorf I, Levy A. P38 Mitogen Activated Protein Kinase alpha (MAPKα) Inhibition to Promote Neurologic Recovery in Rat Stroke Transient Middle Cerebral Artery Occlusion (t-MCAO) Model. Neurology 2016; 86 (Supplement):P5.228.
33. Chollet F. Pharmacologic approaches to cerebral aging and neuroplasticity: insights from the stroke model. Dialogues Clin Neurosci. 2013;1 15:67-76.
34. Gold CA, Budson AE. Memory loss in Alzheimer’s disease: implications for development of therapeutics. Expert Rev Neurother. 2008; 8:1879-1891.
35. Tromp D, Dufour A, Lithfous S, Pebayle T, Despres O. Episodic memory in normal aging and Alzheimer disease: Insights from imaging and behavioral studies. Ageing Res Rev. 2015;2 4:232-262.
36. Scheltens P, Twisk JW, Blesa R, et al. Efficacy of Souvenaid in mild Alzheimer’s disease: results from a randomized, controlled trial. J Alzheimers Dis. 2012;3 1:225-236.
37. Scheltens P, Kamphuis PJ, Verhey FR, et al. Efficacy of a medical food in mild Alzheimer’s disease: A randomized, controlled trial. Alzheimers Dement. 2010; 6:1-10.e11.
38. Phillips MA, Childs CE, Calder PC, Rogers PJ. No Effect of Omega-3 Fatty Acid Supplementation on Cognition and Mood in Individuals with Cognitive Impairment and Probable Alzheimer’s Disease: A Randomised Controlled Trial. Int J Mol Sci. 2015;16:24600-24613.
39. Ozenli Y DY, Karaca S. Efficacy of Donepezil on Cognitive Functions in MildCognitive Impairment. Int Bull Clin Psychopharm 2007; 17:62-67.