
R. Milne1, E. Bunnik2, K. Tromp2, S. Bemelmans3, S. Badger1, D.Gove4, M. Maman5, M. Schermer2, L. Truyen6, C. Brayne1, E. Richard3
1. Institute of Public Health, University of Cambridge, United Kingdom; 2. Erasmus University Medical Centre, Rotterdam, the Netherlands; 3. Radboud University Medical Center, the Netherlands; 4. Alzheimer Europe; 5. Novartis Pharma AG, Basel, Switzerland; 6. Janssen Research & Development LLC.
Corresponding Author: Richard Milne, PhD, Institute of Public Health, University of Cambridge, 2 Wort’s Causeway Cambridge, UK, CB1 8RN, rjm231@cam.ac.uk, 0044 (0)1223 761912
J Prev Alz Dis 2016;in press
Published online February 22, 2017, http://dx.doi.org/10.14283/jpad.2017.5
Abstract
There is growing interest in the development of novel approaches to secondary prevention trials in Alzheimer’s disease to facilitate screening and recruitment of research participants and to reduce the time and costs associated with clinical trials. Several international research collaborations are setting up research infrastructures that link existing research cohorts, studies or patient registries to establish ‘trial-ready’ or ‘readiness’ cohorts. From these cohorts, individuals are recruited into clinical trial platforms. In setting up such research infrastructures, researchers must make ethically challenging design decisions in at least three areas: re-contacting participants in existing research studies, obtaining informed consent for participation in a readiness cohort, and disclosure of Alzheimer’s disease-related biomarkers. These ethical considerations have been examined by a dedicated workgroup within the European Prevention of Alzheimer’s Dementia (EPAD) project, a trans-European longitudinal cohort and adaptive proof-of-concept clinical trial platform. This paper offers recommendations for the ethical management of re-contact, informed consent and risk disclosure which may be of value to other research collaborations in the process of developing readiness cohorts for prevention trials in Alzheimer’s disease and other disease areas.
Key words: Ethics,recruitment,readiness cohort,consent, disclosure, Alzheimer’s disease.
Introduction
No new drugs for Alzheimer’s disease have become available in over a decade, despite significant research and development efforts and a high number of late phase clinical trials (1). This failure has been attributed in part to the choice of study population (1, 2): while the majority of past research has focused on patients with mild-to-moderate Alzheimer’s dementia, recent clinical trials have begun to concentrate on the earlier stages of the disease identified by revised research diagnostic criteria (3, 4). Such studies aim to prevent the development or progression of cognitive impairment among people who do not show any signs of dementia but show biomarker changes thought to be associated with increased risk of developing Alzheimer’s dementia (5–7).
Moreover, methodological issues associated with the definition of Alzheimer’s Disease itself, study recruitment, retention and inter-site variability have been identified as barriers to innovation (8–11). Several public-private research collaborations are currently involved in developing new models for conducting clinical trials in Alzheimer’s disease. These involve the establishment of ‘readiness’ (12) or ‘trial-ready’ cohorts. The primary goal of these longitudinal cohorts is to provide a well-characterised population of potential research participants for recruitment into prevention trials, to limit screening failure rates and to reduce recruitment time. The readiness cohorts themselves may be drawn from existing research registers and cohorts and feed into clinical trial ‘platforms’ (13).
The development of readiness cohorts for clinical trial platforms aimed at altering the natural history of underlying brain changes associated with Alzheimer’s dementia has been described as the ‘pivotal’ (12) element in the approach adopted by a growing number of large public-private initiatives in Alzheimer’s disease research. These include the European Prevention of Alzheimer’s Dementia project (EPAD) (12) and the Global Alzheimer’s Platform (GAP) (13). A simplified model of the approach developed by EPAD and GAP is shown in figure 1. Both projects recruit participants from existing registries and observational cohorts into a new longitudinal cohort study. Participants complete an extensive battery of cognitive tests and biological examinations and are followed over time. This cohort can then function both as a readiness cohort for secondary prevention trials – to be run on a clinical trial platform – and as a resource for disease modelling. Similar models are being adopted within the Dementias Platform UK, and within industry-sponsored studies (14).
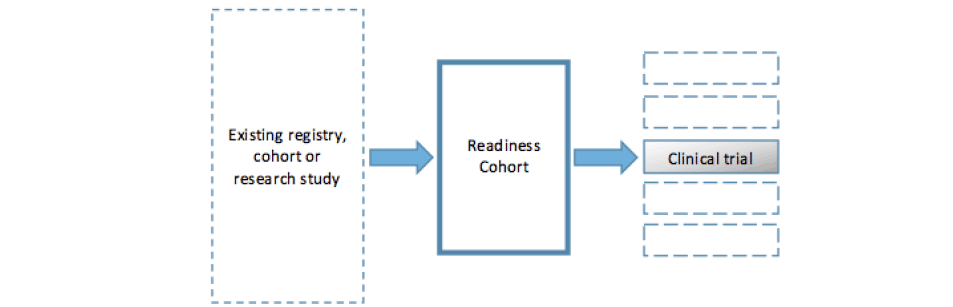
Figure 1. Simplified model of a readiness cohort drawn from existing studies and leading to one or several clinical trials
The development of linked research infrastructures for secondary prevention studies represents an innovative approach to drug development for Alzheimer’s disease. It also raises distinct ethical questions which recombine those traditionally associated with the conduct of cohort studies and clinical trials. These questions include the recruitment of participants from existing studies, ensuring informed consent as participants move through the research process, and the disclosure of Alzheimer’s dementia risk biomarker status.
Ethical issues associated with the design and development of readiness cohorts
All medical research requires careful attention to ethical values and principles. General guidelines, declarations and frameworks exist to guide researchers accordingly (15–17). However, the specific concerns presented by different types of research conducted in different clinical domains may vary significantly. The ethical questions associated with research in Alzheimer’s dementia have long been primarily concerned with the potential risks and burdens of research and the capacity of people with dementia to provide informed consent (18–23). Since the identification of the ApoE susceptibility gene, these concerns has been accompanied by debate around the potential psychological impact on asymptomatic individuals of learning disease risk (24, 25). Most recently, specific attention has been paid to the issues associated with prevention and biomarker-led research in ‘preclinical’ populations (26, 27), in which biomarker levels presumed to reflect Alzheimer’s disease pathology and an assumed increased risk of later dementia are present while cognitive functioning is normal. These issues revolve around the impact on healthy individuals of receiving Alzheimer’s dementia risk information associated with a high degree of uncertainty and with no clear course of action to reduce risk.
In the following sections we identify three areas that require particular attention in the early stages of setting up long-term linked research projects involving readiness cohorts in Alzheimer’s disease research. First, we cover the recruitment of participants from existing research cohorts and registers and the responsibilities of research collaborations in this area. Second, we outline ethical issues related to informed consent and recruitment into multi-stage, linked projects. The third and final area of discussion examines particular challenges associated with the disclosure of risk status within readiness cohorts linked to secondary prevention studies.
Recruiting from existing studies
Existing registries and longitudinal cohort studies can act as resources for the recruitment of well-characterised participants for Alzheimer’s disease research (10). The identification of eligible individuals or groups of participants within existing studies inevitably involves some level of screening of existing studies’ data. There are two pertinent issues in relation to this process. The first relates to how and by whom data is accessed in order to re-contact participants. The second relates to the ability of studies to re-contact and the procedure for doing so.
The screening of data from existing studies may be undertaken by researchers in the new readiness cohort, based on data shared by these existing studies; or it may be carried out by the local cohort investigators based on selection criteria provided by the readiness cohort. If the former approach is adopted, the access, use, processing, and sharing of research participants’ coded (or identifiable) personal health information and research data should adhere to international ethical guidelines, including the draft WMA declaration on ethical considerations regarding health databases and biobanks (28-30). In particular, in any screening process researchers must ensure that no personal data is shared without participants’ consent.
In the second case, existing studies can share only ‘metadata’ which describe the scope (and types) of data available and the number of people on whom they are held. These metadata can be used to establish whether or not the original study contains a substantial population of eligible participants. In such cases, no individual-level data are accessed by the new readiness cohort and consequently no consent from research participants is required. Local cohort investigators can then identify participants who meet the inclusion criteria for the new study and establish whether they are eligible to be re-contacted.
Approaches to re-contact for research by third parties have received comparatively little attention in discussions of biomedical research ethics (31), or within the governance structures of studies themselves. In re-contacting participants, researchers must ensure that participants’ autonomy is respected, that their privacy is protected and that they are not exposed to either unnecessary risks or unacceptable burdens through further participation. These principles apply to research more generally (15, 32), but may be particularly important in a context where individuals are asked to extend their existing participation in a new direction. The specific conditions for re-contact for readiness cohort should be established through discussions between existing studies and their institutional review boards or ethics committees. When consent for re-contact is not in place, investigators of the original study should first ask their participants to consent to being re-contacted about further studies. Only those who have consented can then be contacted about particular further studies. A decision flow for this process is shown in figure 2 below.
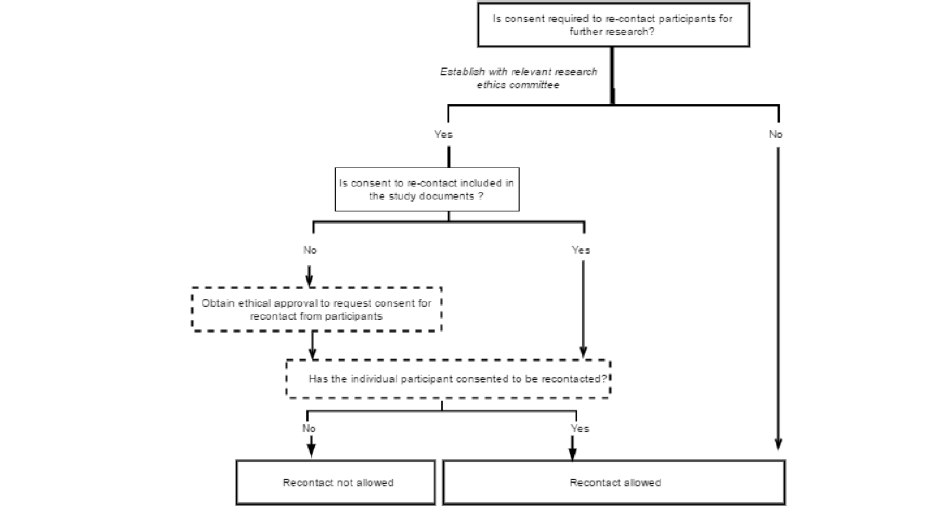
Figure 2. Recontact flow chart
If participants are eligible to be re-contacted, the investigators of the original study can provide information about the new study and ask selected participants to contact the investigators of the new study if they are interested in taking part. In this approach, participants are asked to opt-in to taking part in the new study.
Finally, it is important to recognise that new readiness cohorts and other initiatives which draw on existing studies have a responsibility to minimise any wider negative impact. There is a risk that participation in a readiness cohort may increase drop-out rates from existing studies, particularly if taking part is a comparatively intensive experience. The existence of any such effect should be assessed as readiness cohorts develop, and researchers should involve working closely with investigators and participants in existing studies to ensure that the prior commitments and choices of the latter are not counteracted or frustrated. This also implies that individuals who decide to take part in the new readiness cohort should not be excluded from the original study through which they are contacted.
Ensuring informed consent
Readiness cohorts represent one stage on a research journey that may start in a clinical registry or observational cohort and end with participation in a clinical trial. In this respect they differ from traditional longitudinal cohorts or traditional clinical trials, as they extend over time and encompass various stages in which consent may be asked from participants by different parties and for different purposes.
The transitional nature of readiness cohorts presents a distinct challenge when it comes to ensuring that potential participants are fully informed about the scope of the project before consenting to take part. The requirement of informed consent aims to ensure that research participation is the result of autonomous choice (32, 33). Informed consent is given to a specific party or person for a specific task or activity, and research participants must be made aware of what is involved in this task, including the risks, potential benefits and burden of participation, and must choose voluntarily to take part. However, while full, relevant, and accurate information about a research project is indispensable (34) in order to give informed consent, this information may not be available in readiness cohorts, where the various stages of a project are distinct but interconnected, and where it is not clear what the final journey of any individual participant through the project will be. After all, participants in a readiness cohort do not know at the outset whether or not they will eventually be asked to participate in a clinical trial, and what that trial will entail.
The ethical concern in this context is that by only providing information about the specific activities for which informed consent is sought at a particular point in time, readiness cohorts would risk becoming ‘fish traps’ (35, 36) for their participants (see figure 3). When a participant initially consented to take part in the original registry or cohort, they did not consent to the tests and examinations of the new readiness cohort. Or when a participant initially consented to being re-contacted about further research studies, they did not consent to undergoing a lumbar puncture as part of the new readiness cohort, to learning about their Alzheimer’s disease risk, or taking drugs in the context of a clinical trial. With each step, the burdens and risks of research participation will accumulate incrementally. At the same time, once started on the path of participation, it may become progressively more difficult to withdraw. This ‘luring’ or ‘easing’ of participants into clinical trial participation would be a form of misleading or deceiving participants, which would run counter to the ethical requirement of informed consent. It may also increase the risk of drop-out, jeopardising the scientific integrity of the data for the purpose of natural disease course monitoring by introducing additional bias.
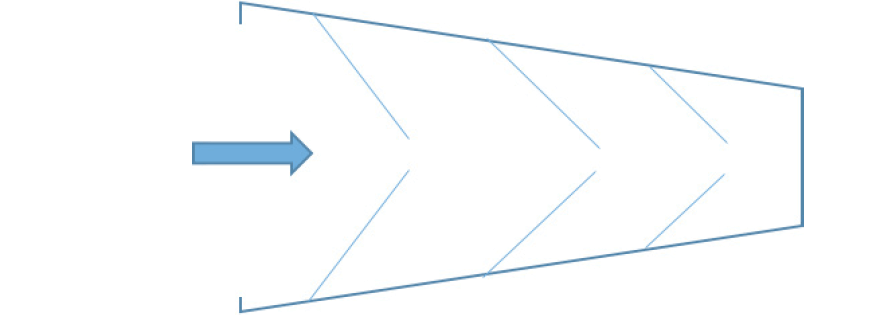
Figure 3. The fish trap
The concept of the fish trap has been described in the context of prenatal screening (35, 36) where healthy pregnant women are not confronted with difficult reproductive decisions in a single stroke, but progressively, step-by-step. When they first consent to participate in screening, they do not (yet) consent to the steps that may follow – amniocentesis or chorionic villus sampling, and eventually termination of the pregnancy. With each step, however, it becomes more difficult to turn back.
In order to prevent a fish trap, global information about the whole study trajectory must be available to potential participants from the outset. A staged model for informed consent (37) should present information about the entire study journey to research participants at every step. Informed consent requires information specific to the stage for which consent is sought. At each step of the way, however, participants should also be informed about further stages of the research project and the project in its entirety. A staged consent approach allows for study information to be repeated, rehearsed and built upon and for participants to be reminded that they are able to withdraw at any time without having to give any reasons. Without this general information, consent would not be informed consent. A simplified version of the staged model for informed consent that was adopted in EPAD is shown in figure 4 below.
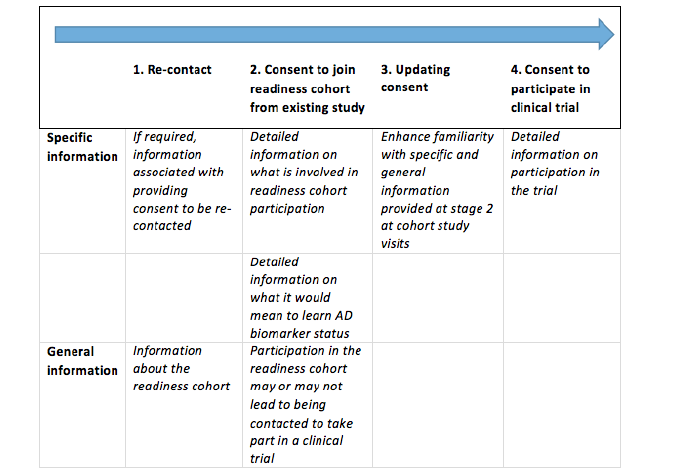
Figure 4. The staged consent model adopted in EPAD
Education and risk disclosure
A core ethical consideration associated with secondary prevention research in Alzheimer’s disease relates to the potential impact on research participants of learning that one is at elevated risk of developing Alzheimer’s dementia, based on either ApoE genotype or other biomarkers, such as beta-amyloid status (26, 38). Concerns about disclosure of ApoE (39) and amyloid status (40, 41) revolve around the potential the psychological, behavioural and social harms of disclosure, particularly of amyloid status (42).
In the case of readiness cohort, decisions about whether or not to disclose risk information are encountered at three stages: during contact to take part in a readiness cohort; following cohort examinations and assessments; and as part of the recruitment from readiness cohorts into clinical trials. At each stage, the pros and cons of disclosure differ.
At the first stage, that of contact to take part in the readiness cohort, the decision over whether or not to disclose risk status is shaped by the proposed constituency of the readiness cohort. If a readiness cohort aims to recruit only a population thought to be at increased risk of developing Alzheimer’s dementia, disclosure is essential in order to make individuals aware why they are considered eligible for the research – a well-established research-ethical requirement (32). If, in contrast, the readiness cohort includes a wide range of risk states and researchers are blinded to why any individual is contacted, it is no longer clear why any individual participant has been contacted. However, the inclusion of ‘lower risk’ participants should not occur solely as a means of avoiding disclosure, not least because these participants would be subjected to potentially unnecessary risks and burdens as a consequence.
At the second stage, guidance frameworks for population-based research suggest that individual research results should be returned if consent is in place, if the findings are analytically valid, if they reveal a significant risk of a serious health condition, and finally if they are actionable (43). Neither ApoE nor amyloid information has confirmed clinical utility and robust information about likely disease progression in individuals does not yet exist (44). However, this may be changing (45) and indeed risk information may be of value to research participants regardless of the lack of effective treatments (46).
Finally, ApoE and amyloid status should be disclosed as part of a transparent enrolment process to secondary prevention clinical trials (47), as is the case in a number of ongoing studies (26, 48, 49). This reflects the requirement to make individuals aware of why they are considered eligible for research. As such, participants in readiness cohorts should be informed on what basis they are contacted for any clinical trial. However, there are important features of this process which differ from conventional clinical trial structures.
Within recruitment to conventional trial structures, participants can be provided with information at the point of screening for the study to enable them to decide whether or not risk information is something they would like to know (48, 50). In the context of readiness cohorts, participants are enrolled at a stage when it is not yet known whether or not they will participate in a trial or for what reasons (i.e. based on what biomarkers) they may be recruited. However, because participants are aware that the cohort exists to facilitate trials in preclinical/at-risk and prodromal Alzheimer’s disease populations, being invited to take part in a trial necessarily constitutes disclosure of preclinical/at-risk or prodromal status. To choose not to know this information would be to choose not to be contacted for a trial, which is incompatible with the primary goal of a readiness cohort.
The complexities associated with disclosing risk status within linked research structures prompt three core recommendations for establishing readiness cohorts.
Firstly, all participants entering readiness cohorts should be willing to learn biomarker results related to their risk of developing Alzheimer’s dementia and understand the uncertainty of that evidence, as there is no effective way of protecting an individual’s right not to know (this information) while preserving transparent enrolment procedures.
Secondly, even if no return of individual research results will occur during the cohort stage of research, the link to secondary prevention studies means that all potential participants should receive educational materials and/or briefings about risk information during the cohort consent process. This ensures that potential participants are informed about what biomarkers can and cannot tell them about their risk of developing Alzheimer’s dementia, and allows them to make an informed decision about whether this is information they would like to learn in the future.
Finally, readiness cohort researchers should work with trial sponsors to ensure that the disclosure of biomarker information from cohort examinations is carried out by appropriately qualified professionals.
Conclusion
The development of linked projects for Alzheimer’s disease research, which draw on existing research studies to establish readiness cohorts for secondary prevention clinical trials, raises distinct ethical challenges. These derive from the novelty of the approach and how it combines and reframes existing issues of concern. Particular ethical considerations and challenges revolve around the process of identifying and contacting potential participants in the readiness cohort; the staged provision of specific and general information to research participants in order to prevent them entering a ‘fish trap’, and the establishment of robust procedures for informing participants about the meaning of risk information and for disclosing risk. Recommendations in these areas are summarised in Box 1.
The concerns and recommendations detailed here suggest that it is critically important that researchers consider the journey of a research participant through the various stages of the research project from start to finish, from re-contact to clinical trial participation. This includes considering how challenges associated with later stages of research, such as recruitment from a readiness cohort into a clinical trial which may involve risk disclosure, affect the information that should be provided to participants during initial recruitment into the cohort. It is critical to the success of readiness cohorts, however, to carefully align re-contact, informed consent and risk disclosure processes throughout the research project.

Box 1. Recommendations
Funding: This work was funded through the Ethical Legal and Social Implications work package of the European Prevention of Alzheimer’s Dementia (EPAD) study EPAD receives support from the Innovative Medicines Initiative Joint Undertaking under grant agreement n° 115736, resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007-2013) and EFPIA companies’ in kind contribution. RM was also funded through the UK National Institute of Health Research grant to the Cambridge Biomedical Research Centre.
Acknowledgements: We are grateful to members of the EPAD Ethics Advisory board, Jason Karlawish, Marianne Boenink, Xavier Carne and David Good for their comments on the discussions summarised in this paper. We would also like to thank members of the EPAD consortium for discussion and comments related to the development of these recommendations. All authors contributed to the conception and development of the manuscript, have either drafted or critically revised the content, and have approved the final version of the manuscript.
Conflict of interest: LT is an employee of Janssen R&D LLC, EFPIA co-lead of the IMI-EPAD project and member of the Johnson&Johnson BioResearch Ethics Committee. MM is an employee of Novartis Pharma AG. The opinions expressed in this article are those of the authors and do not necessarily reflect the views of their employers or organizations. RM, EB, KT, SBe, SBa, DG, MS, CB and ER have no conflicts of interest with this paper.
Ethical stantards: N/A
References
1. Schneider LS, Mangialasche F, Andreasen N, Feldman H, Giacobini E, Jones R, et al. Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. J Intern Med. 2014;275: 251–83. doi:10.1111/joim.12191
2. Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6: 37. doi:10.1186/alzrt269
3. Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7: 280–92. doi:10.1016/j.jalz.2011.03.003
4. Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 2014;13: 614–29. doi:10.1016/S1474-4422(14)70090-0
5. Carrillo MC, Brashear HR, Logovinsky V, Ryan JM, Feldman HH, Siemers ER, et al. Can we prevent Alzheimer’s disease? Secondary “prevention” trials in Alzheimer’s disease. Alzheimer’s Dement. 2013;9: 123–131.e1.
6. Vellas B, Aisen PS, Sampaio C, Carrillo M, Scheltens P, Scherrer B, et al. Prevention trials in Alzheimer’s disease: An EU-US task force report. Prog Neurobiol. 2011;95: 594–600.
7. Sperling R, Mormino E, Johnson K. The evolution of preclinical Alzheimer’s disease: implications for prevention trials. Neuron. Elsevier; 2014;84: 608–22. doi:10.1016/j.neuron.2014.10.038
8. Vellas B, Hampel H, Rougé-Bugat ME, Grundman M, Andrieu S, Abu-Shakra S, et al. Alzheimer’s disease therapeutic trials: EU/US Task Force report on recruitment, retention, and methodology. J Nutr Health Aging. 2012;16: 339–45.
9. Feldman HH, Haas M, Gandy S, Schoepp DD, Cross AJ, Mayeux R, et al. Alzheimer’s disease research and development: a call for a new research roadmap. Ann N Y Acad Sci. 2014;1313: 1–16. doi:10.1111/nyas.12424
10. Aisen P, Touchon J, Andrieu S, Boada M, Doody R, Nosheny RL, et al. Registries and cohorts to accelerate early phase alzheimer’s trials. A report from the E.U/U.S. clinical trials in Alzheimer’s disease task force. J Prev Alzheimer’s Dis. 2016;3: 68–74.
11. Ritchie CW, Terrera GM, Quinn TJ. Dementia trials and dementia tribulations: methodological and analytical challenges in dementia research. Alzheimers Res Ther. 2015;7: 31. doi:10.1186/s13195-015-0113-6
12. Ritchie CW, Molinuevo JL, Truyen L, Satlin A, Van der Geyten S, Lovestone S. Development of interventions for the secondary prevention of Alzheimer’s dementia: the European Prevention of Alzheimer’s Dementia (EPAD) project. The Lancet Psychiatry. Elsevier; 2015;3: 179–186. doi:10.1016/S2215-0366(15)00454-X
13. Cummings J, Aisen P, Barton R, Bork J, Doody R, Dwyer J, et al. Re-Engineering Alzheimer Clinical Trials: Global Alzheimer’s Platform Network. J Prev Alzheimer’s Dis. 2016;3: 114–120.
14. Janssen. NCT02114372: Cognitive Health in Ageing Register: Investigational, Observational and Trial Studies in Dementia Research: Prospective Readiness Cohort Study (CHARIOT:PRO). 2016. Available: https://clinicaltrials.gov/ct2/show/NCT02114372
15. US Department of Health and Human Services. The Belmont report: Ethical principles and guidelines for the protection of human subjects of research. 1978;
16. Beauchamp T, Childress J. Principles of biomedical ethics. Oxford: Oxford University Press; 2001.
17. World Medical Association. Declaration of Helsinki. Ethical principles for medical research involving human subjects. 2013;
18. Melnick VL, Dubler NN, Weisbard A, Butler RN. Clinical Research in Senile Dementia of the Alzheimer Type. J Am Geriatr Soc.; 1984;32: 531–536. doi:10.1111/j.1532-5415.1984.tb02240.x
19. Whitehouse PJ. Future prospects for Alzheimer’s disease therapy: ethical and policy issues for the international community. Acta Neurol Scand.; 1996;94: 145–149. doi:10.1111/j.1600-0404.1996.tb05885.x
20. Karlawish JH, Casarett D. Addressing the ethical challenges of clinical trials that involve patients with dementia. J Geriatr Psychiatry Neurol. 2001;14: 222–8.
21. Kim SYH. The ethics of informed consent in Alzheimer disease research. Nat Rev Neurol. 2011;7: 410–4. doi:10.1038/nrneurol.2011.76
22. Nuffield Council on Bioethics. Dementia: Ethical Issues. London; 2009.
23. National Bioethics Advisory Commission. Research Involving Persons With Mental Disorders. Report and Recommendations of the National Bioethics Advisory Commission. Washington; 1998.
24. High DM, Whitehouse PJ, Post SG, Berg L. Guidelines for addressing ethical and legal issues in Alzheimer disease research: a position paper. Alzheimer Dis Assoc Disord. 1994;8: 66–74.
25. Post SG, Whitehouse PJ, Binstock RH, Bird TD, Eckert SK, Farrer LA, et al. The clinical introduction of genetic testing for Alzheimer disease. An ethical perspective. JAMA. 1997;277: 832–6.
26. Molinuevo JL, Cami J, Carné X, Carrillo MC, Georges J, Isaac MB, et al. Ethical challenges in preclinical Alzheimer’s disease observational studies and trials: Results of the Barcelona summit. Alzheimers Dement. 2016; doi:10.1016/j.jalz.2016.01.009
27. Karlawish J. Addressing the ethical, policy, and social challenges of preclinical Alzheimer disease. Neurology. 2011;77: 1487–93. doi:10.1212/WNL.0b013e318232ac1a
28. World Medical Association. Declaration On Ethical Considerations Regarding Health Databases And Biobanks (draft). 2015.
29. Nuffield Council on Bioethics. The collection, linking and use of data in biomedical research and health care: ethical issues. London; 2015.
30. Knoppers B. Framework for responsible sharing of genomic and health-related data. Hugo J. Springer; 2014;8: 3. doi:10.1186/s11568-014-0003-1
31. Beskow LM, Fullerton SM, Namey EE, Nelson DK, Davis AM, Wilfond BS. Recommendations for ethical approaches to genotype-driven research recruitment. Hum Genet. NIH Public Access; 2012;131: 1423–31. doi:10.1007/s00439-012-1177-z
32. Council for International Organizations of Medical Sciences. International Ethical Guidelines for Biomedical Research Involving Human Subjects.. Bulletin of Medical Ethics. 2002.
33. Manson NC, O’Neill O. Rethinking Informed Consent in Bioethics. Cambridge: Cambridge University Press; 2007.
34. Faden R, Beauchamp T. A history and theory of informed consent. Oxford: Oxford University Press; 1986.
35. de Wert G, Dondorp W. Ethiek van voortplantingsgeneeskunde. In: Heineman MJ, Evers JLH, Massuger LFAG, Steegers EAP, editors. Obstetrie en gynaecologie. Houten: Bohn Stafleu van Loghum; 2012. pp. 19–64. doi:10.1007/978-90-368-1191-0
36. Jong A de. Prenatal screening à la carte? 2013; Unpublished PhD thesis. Available: http://digitalarchive.maastrichtuniversity.nl/fedora/get/guid:5834a635-31e5-473a-ac87-e21e1068266b/ASSET1
37. Bunnik EM, Janssens ACJW, Schermer MHN. A tiered-layered-staged model for informed consent in personal genome testing. Eur J Hum Genet. Nature Publishing Group; 2013;21: 596–601. doi:10.1038/ejhg.2012.237
38. Sperling RA, Karlawish J, Johnson KA. Preclinical Alzheimer disease-the challenges ahead. Nat Rev Neurol. 2013;9: 54–8. doi:10.1038/nrneurol.2012.241
39. Goldman JS, Hahn SE, Catania JW, LaRusse-Eckert S, Butson MB, Rumbaugh M, et al. Genetic counseling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet Med. 2011;13: 597–605. doi:10.1097/GIM.0b013e31821d69b8
40. Johnson KA, Minoshima S, Bohnen NI, Donohoe KJ, Foster NL, Herscovitch P, et al. Appropriate use criteria for amyloid PET: a report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association. Alzheimers Dement. 2013;9: e–1–16. doi:10.1016/j.jalz.2013.01.002
41. Leuzy A, Zimmer ER, Heurling K, Rosa-Neto P, Gauthier S. Use of amyloid PET across the spectrum of Alzheimer’s disease: clinical utility and associated ethical issues. Amyloid. Informa UK Ltd. London; 2014;21: 143–8. doi:10.3109/13506129.2014.926267
42. Bemelmans, S., K. Tromp, E. Bunnik, R. Milne, S. Badger, C. Brayne, M. Schermer, and E. Richard. 2016. “Psychological, Behavioral and Social Effects of Disclosing Alzheimer’s Disease Biomarkers to Research Participants – a Systematic Review.” Alzheimer’s Research & Therapy. 8:46 DOI: 10.1186/s13195-016-0212-z
43. Knoppers BM, Deschênes M, Zawati MH, Tassé AM. Population studies: return of research results and incidental findings Policy Statement. Eur J Hum Genet. 2013;21: 245–7. doi:10.1038/ejhg.2012.152
44. Noel-Storr AH, Flicker L, Ritchie CW, Nguyen GH, Gupta T, Wood P, et al. Systematic review of the body of evidence for the use of biomarkers in the diagnosis of dementia. Alzheimers Dement. 2013;9: e96–e105. doi:10.1016/j.jalz.2012.01.014
45. Lingler JH, Klunk WE. Disclosure of amyloid imaging results to research participants: Has the time come? Alzheimer’s Dement. NIH Public Access; 2013;9: 741–744.e2. doi:10.1016/j.jalz.2012.09.014
46. Gooblar J, Roe CM, Selsor NJ, Gabel MJ, Morris JC. Attitudes of Research Participants and the General Public Regarding Disclosure of Alzheimer Disease Research Results. JAMA Neurol. 2015; 1. doi:10.1001/jamaneurol.2015.2875
47. Kim SYH, Karlawish J, Berkman BE. Ethics of genetic and biomarker test disclosures in neurodegenerative disease prevention trials. Neurology. 2015; WNL.0000000000001451–. doi:10.1212/WNL.0000000000001451
48. Harkins K, Sankar P, Sperling R, Grill JD, Green RC, Johnson KA, et al. Development of a process to disclose amyloid imaging results to cognitively normal older adult research participants. Alzheimers Res Ther. 2015;7: 26. doi:10.1186/s13195-015-0112-7
49. Tariot PN, Ho C, Langlois C, Reiman EM, Lopera F, Langbaum JB, et al. The Alzheimer’s Prevention Initiative. Alzheimer’s Dement. 2014;10: P247. doi:10.1016/j.jalz.2014.04.379
50. Lingler JH, Butters MA, Gentry AL, Hu L, Hunsaker AE, Klunk WE, et al. Development of a Standardized Approach to Disclosing Amyloid Imaging Research Results in Mild Cognitive Impairment. J Alzheimers Dis. 2016;52: 17–24. doi:10.3233/JAD-150985