
P.S. Aisen1, J. Cummings2, R. Doody3, L. Kramer4, S. Salloway5, D.J. Selkoe6, J. Sims7, R.A. Sperling6, B. Vellas8 and the EU/US CTAD 2019 Task Force*
* EU/US/CTAD TASK FORCE: Susan Abushakra (Framingham) ; Paul Aisen (San Diego) ; John Alam (Boston) ; Sandrine Andrieu (Toulouse) ; Anu Bansal (Simsbury) ; Monika Baudler (Basel) ; Joanne Bell (Wilmington) ; Mickaël Beraud (Zaventem); Tobias Bittner (Basel); Samantha Budd Haeberlein (Cambridge) ; Szofia Bullain (Basel) ; Marc Cantillon (Gilbert) ; Maria Carrillo (Chicago) ; Carmen Castrillo-Viguera (Cambridge) ; Ivan Cheung (Woodcliff Lake) ; Julia Coelho (San Francisco) ; Jeffrey Cummings (Las Vegas) ; Michael Detke (San Francisco) ; Daniel Di Giusto (Basel) ; Rachelle Doody (South San Francisco) ; John Dwyer (Washington) ; Michael Egan (North Wales) ; Colin Ewen (Slough) ; Charles Fisher (San Francisco) ; Serge Gauthier (Montreal) ; Michael Gold (North Chicago) ; Harald Hampel (Woodcliff Lake) ; Ping He (Cambridge) ; Suzanne Hendrix (Salt Lake City) ; David Henley (Titusville) ; Michael Irizarry (Woodcliff Lake) ; Atsushi Iwata (Tokyo) ; Takeshi Iwatsubo (Tokyo) ; Michael Keeley (South San Francisco) ; Geoffrey Kerchner (South San Francisco) ; Gene Kinney (San Francisco) ; Hartmuth Kolb (Titusville) ; Marie Kosco-Vilbois (Lausanne) ; Lynn Kramer (Westport) ; Ricky Kurzman (Woodcliff Lake) ; Lars Lannfelt (Uppsala) ; John Lawson (Malvern) ; Jinhe Li (Gilbert) ; Frank Longo (Stanford) ; Mark Mintun (Philadelphia) ; Vaidrius Navikas (Valby) ; Gerald Novak (Titusville) ; Gunilla Osswald (Stockholm) ; Susanne Ostrowitzki (South San Francisco) ; Anton Porsteinsson (Rochester) ; Rema Raman (San Diego) ; Ivana Rubino (Cambridge) ; Marwan Sabbagh (Las Vegas) ; Stephen Salloway (Providence) ; Rachel Schindler (New York) ; Lon Schneider (Los Angeles) ; Hiroshi Sekiya (Malvern) ; Dennis Selkoe (Boston) ; Eric Siemers (Zionsville) ; John Sims (Indianapolis) ; Lisa Sipe (San Marcos) ; Olivier Sol (Lausanne) ; Reisa Sperling (Boston) ; Andrew Stephens (Berlin) ; Johannes Streffer (Braine-l’Alleud) ; Joyce Suhy (Newark) ; Chad Swanson (Woodcliff Lake) ; Gilles Tamagnan (New Haven) ; Rudolph Tanzi (Boston) ; Pierre Tariot (Phoenix); Edmond Teng (South San Francisco) ; Martin Tolar (Framingham) ; Jacques Touchon (Montpellier) ; Martin Traber (Basel) ; Bruno Vellas (Toulouse) ; Andrea Vergallo (Woodcliff Lake) ; Christian Von Hehn (Cambridge) ; George Vradenburg (Washington) ; Judy Walker (Singapore) ; Michael Weiner (San Francisco) ; Glen Wunderlich (Ridgefield) ; Jennifer Ann Zimmeri (Indianapolis) ; Haichen Yang (North Wales) ; Wagner Zago (San Francisco) ; Thomas Zoda (Austin)
1. Alzheimer’s Therapeutic Research Institute (ATRI), Keck School of Medicine, University of Southern California, San Diego, CA, USA; 2. Department of Brain Health, School of Integrated Health Sciences, University of Nevada Las Vegas, and Cleveland Clinic Lou Ruvo Center for Brain Health, Las Vegas, Nevada, USA; 3. Genentech/Roche, Basel, Switzerland; 4. Eisai Co., Ltd., Eisai, Inc., Woodcliff Lake, NJ, USA; 5. The Warren Alpert Medical School of Brown University, Providence RI, USA; 6. Brigham and Women’s Hospital, Boston MA, USA; 7. Eli Lilly and Company, Indianapolis, IN, USA; 8. Gerontopole, INSERM U1027, Alzheimer’s Disease Research and Clinical Center, Toulouse University Hospital, Toulouse, France
Corresponding Author: P.S. Aisen, University of Southern California Alzheimer’s Therapeutic Research Institute, San Diego, CA, USA, paisen@usc.edu
J Prev Alz Dis 2020;3(7):146-151
Published online April 17, 2020, http://dx.doi.org/10.14283/jpad.2020.24
Abstract
The termination of many clinical trials of amyloid-targeting therapies for the treatment of Alzheimer’s disease (AD) has had a major impact on the AD clinical research enterprise. However, positive signals in recent studies have reinvigorated support for the amyloid hypothesis and amyloid-targeting strategies. In December 2019, the EU-US Clinical Trials on Alzheimer’s Disease (CTAD) Task Force met to share learnings from these studies in order to inform future trials and promote the development of effective AD treatments. Critical factors that have emerged in studies of anti-amyloid monoclonal antibody therapies include developing a better understanding of the specific amyloid species targeted by different antibodies, advancing our insight into the mechanism by which those antibodies may reduce pathology, implementing more comprehensive repertoires of biomarkers into trials, and identifying appropriate doses. Studies suggest that Amyloid-Related Imaging Abnormalities – effusion type (ARIA-E) are a manageable safety concern and that caution should be exercised before terminating studies based on interim analyses. The Task Force concluded that opportunities for developing effective treatments include developing new biomarkers, intervening in early stages of disease, and use of combination therapies.
Key words: Alzheimer’s disease, dementia, amyloid hypothesis, monoclonal antibody treatment, BACE inhibitors, combination therapy.
Introduction
Despite encouraging results from the aducanumab Phase 1 and BAN2401 Phase 2 anti-amyloid antibody clinical trials, amyloid-beta protein (Aß)-based strategies for the treatment of Alzheimer’s disease (AD) appeared to take a crippling blow in March 2019 when Biogen announced it was terminating two clinical trials (EMERGE and ENGAGE) of the anti-Aß monoclonal antibody aducanumab based on the results of an interim analysis demonstrating a lack of benefit or ‘futility.’ The field had another major challenge in July when Novartis, Amgen, and the Banner Alzheimer’s Institute announced termination of pivotal trials of the beta-site amyloid precursor protein cleaving enzyme (BACE) inhibitor umibecestat after an interim analysis identified cognitive worsening in trial participants. This marked the fifth failed BACE inhibitor in less than two years, two with trials stopped because of adverse events (Merck’s verubecestat and Janssen’s atabecestat) and two trials stopped for lack of efficacy (Astra Zeneca and Eli Lilly’s lanabecestat and Eli Lilly’s LY3202626) (1–3). A fifth BACE inhibitor trial of Eisai and Biogen’s elenbecestat was halted in September 2019 due to an unfavorable risk/benefit profile (4). Trials for another anti- Aß monoclonal antibody, Genentech and Roche’s crenezumab, were terminated in 2019 for futility (5).
Then, in October, a stunning reversal: Biogen announced that the futility analysis in the aducanumab trial was misleading. Analysis of a larger data set indicated that aducanumab did indeed slow cognitive decline in trial participants who received a higher dose of the drug for longer periods of time in one of the two studies. Following this announcement, Biogen indicated they planned to submit aducanumab to the U.S. Food and Drug Administration (FDA) for regulatory approval. Any form of approval for aducanumab has the potential to transform the AD field, providing hope for patients and researchers alike. Regulatory success could also reinvigorate support for the amyloid cascade hypothesis, which posits that deposition of Aβ in the brain leads to the neurodegeneration and dementia that characterize AD. This hypothesis has driven the development of AD therapeutics for decades. Secretase inhibitors block production of Aβ, while anti-Aβ antibodies are designed to clear Aβ and prevent the formation of amyloid plaques as well as neutralize soluble Aß oligomers. Prior to the announcement of aducanumab’s potential beneficial effects, no secretase inhibitor and only two monoclonal antibodies — BAN2401 and gantenerumab — had preliminary evidence of possible efficacy against Aß, and there was much speculation in the field that the amyloid hypothesis was dead or at least unhelpful in guiding development of AD therapeutics. However substantial emerging evidence supports the amyloid cascade hypothesis (6).
To better understand the implications of these clinical trial results and the future of amyloid-based therapies, the European Union and United States Clinical Trials on Alzheimer’s Disease Task Force (EU/US CTAD-TF) convened a meeting in San Diego on December 4, 2019, bringing together industry scientists involved in clinical trials of anti- Aß and other AD therapies along with representatives from pharmaceutical, biotechnology, diagnostics, and medical device companies, academic researchers, clinicians, and non-profit organizations. Their goal was to articulate lessons learned from these trials with the hope of enabling future successful trials that will lead to the approval of effective treatments for AD.
Learnings from trials of anti-amyloid monoclonal antibody trials
The Task Force discussed five anti-amyloid monoclonal antibody therapies currently in clinical development: aducanumab (7), BAN2401 (6), gantenerumab (8), solanezumab (9–13) , and donanemab. Other anti-amyloid monoclonal antibodies (e.g., crenezumab) are also in development (5, 14). As summarized in Table 1, these antibodies target different forms of amyloid, may have different mechanisms of action, and are being tested for efficacy at different stages of disease.
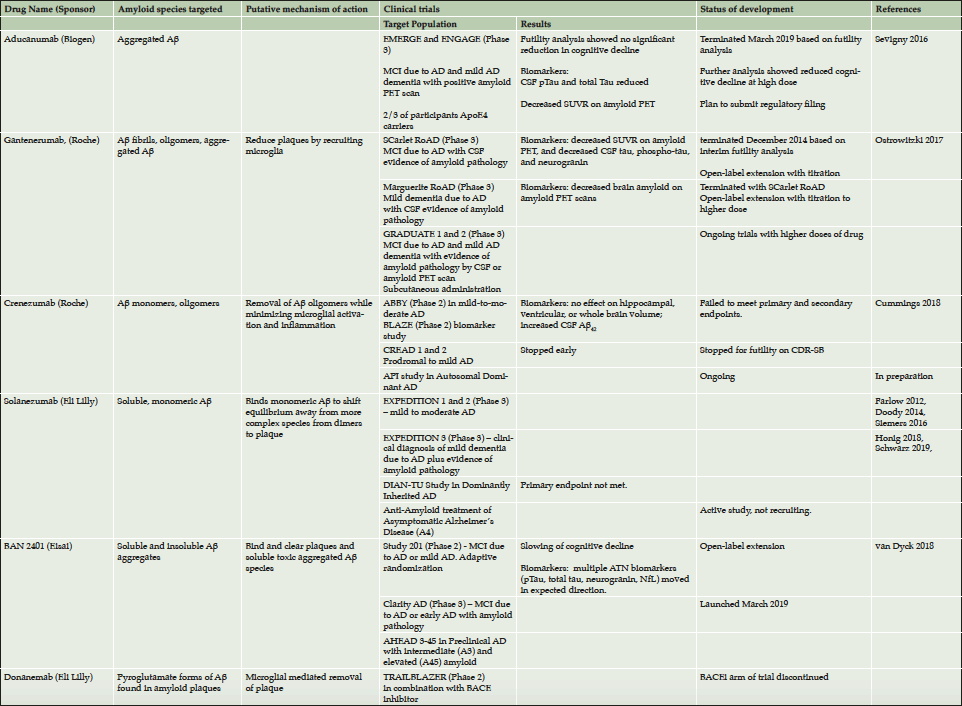
Table 1. Anti-amyloid monoclonal antibody therapies
The importance of dose
The futility analysis in the ENGAGE and EMERGE aducanumab trials – two identically designed Phase 3 studies — was based on a pooled interim dataset of approximately 50% of enrolled participants using a probability calculation that assumed non-heterogeneity between the two studies. A subsequent analysis of a larger dataset, however, revealed that protocol amendments allowing increased dosing in apolipoprotein E epsilon 4 (APOE4) carriers had differential effects on the two studies due to the relative timing of enrollment. This analysis demonstrated a statistically significant reduction in clinical decline across multiple clinical endpoints among early AD patients in EMERGE, likely due to high dose exposure to the drug. Participants in the ENGAGE trial who had received higher doses (10 mg/kg) for at least 10 doses had clinical effects similar to those of the EMERGE participants. Amyloid positron emission tomography (PET) studies demonstrated dose-dependent reduction of brain amyloid deposition across both trials.
Other trials have also demonstrated substantial dose-related amyloid lowering. Study 201 of BAN2401 used an adaptive randomization design with six arms to understand the impact of dose and minimize the number of participants treated with ineffective doses. The highest dose (10 mg/kg biweekly) produced the greatest slowing of disease progression and most robust reduction in brain amyloid levels compared to placebo and is used in the recently-launched Phase 3 Clarity AD study.
Open-label extensions of two early Phase 3 gantenerumab trials, in which study participants were assigned one of five titration schemes, also showed that five times higher dose of ganternerumab than was used in the earlier phase 3 studies drove increased amyloid reduction assessed with amyloid PET imaging (15). These findings prompted the initiation of a new Phase 3 program using this five-fold higher doses.
Mechanism matters
Amyloid is not a monolithic target but a family of monomers, oligomers, protofibrils, and fibrils; and different anti-Aβ antibodies target partially different species. The molecular dynamics by which targeting different species results in variable effects on plaque burden and brain volume loss are not well understood; however, these differential mechanisms may help explain the different trial effects observed.
Solanezumab was hypothesized to remove brain amyloid through what is called the “peripheral sink hypothesis,” i.e., by increasing the clearance of soluble Aβ via the formation of antibody-Aβ complexes in the plasma. However, pharmacodynamic studies showed that a reduction of Aβ in the peripheral compartment failed to shift the equilibrium between Aβ species enough to cause a substantial reduction of fibrillary Aβ in the brain (16); the possible beneficial effect of solanezumab on cognitive decline may nonetheless be mediated by its binding to smaller, diffusible forms. Other possible mechanisms of anti-Aβ antibodies include direct targeting of Aβ plaques or other toxic species of Aβ for removal or activating phagocytosis of Aβ by microglia (17). Clinical trials of solanezumab in mild-moderate AD and in prodromal/mild AD failed to show a drug-placebo difference and no effects on biomarkers were observed. Solanezumab continues in the Anti-Amyloid treatment of Asymptomatic Alzheimer’s disease (A4) study of cognitively asymptomatic participants with positive amyloid imaging.
The effects of anti-Aβ antibodies on brain volume loss is poorly understood. In the EXPEDITION trials, treatment with solanezumab showed a modest but statistically insignificant slowing of brain atrophy (13). Gantenerumab produced no such effects on the measures collected (8). One theory suggests that driving down amyloid may itself be reflected as a reduction in brain volume. The effects on brain volume, however, could differ depending on which form of amyloid the antibody targets (e.g. plaques versus oligomeric forms). Further analysis of data from anti-Aβ antibody trials may help clarify this issue. The correlation of treatment-related brain volume loss and disease progression is also unclear.
ARIA appears to be a manageable safety concern
The incidence of amyloid-related imaging abnormalities – effusion type (ARIA-E) associated with anti-Aβ antibody treatment has been a substantial concern in the development of these therapies (18). For example, in the aducanumab trials, ARIA-E was seen in more than one-third of participants, although these episodes were typically asymptomatic and resolved within 4-16 weeks without long-term sequelae. ARIA-E was also observed in about 10% of participants in the BAN2401 Phase 2 study, occurring primarily in the first three months of treatment.
Recent studies suggest that ARIA-E can be safely managed by titrating drug to the target dose. For example, in the gantenerumab studies, titrating to the target dose reduced ARIA-E incidence in both APOE4 carriers and non-carriers and the majority of episodes were asymptomatic. Other studies have suggested that APOE4 carriers are at higher risk for ARIA-E. While ARIA-E appears to be manageable, uncertainty remains about whether even a minimal risk could be problematic for preclinical AD patients, or whether ARIA-E occurs less frequently in earlier stages of disease or in individuals with lower levels of vascular amyloid.
Although it may be challenging, it will be necessary to develop criteria that could be used in primary care settings for safely beginning treatment and monitoring for ARIA-E should an anti-Aβ monoclonal antibody treatment be approved for AD,. A better understanding of the mechanisms involved could relieve concerns among primary care physicians once these therapies become available.
Interim and futility analyses are useful only if appropriately designed
Futility analyses are designed to protect participants from unnecessary exposure to drugs that have little chance of providing benefits, but if they result in premature termination of a trial, participants and sponsors alike – indeed, the entire field – may suffer adverse consequences from a failure to identify efficacious treatments and the failure to collect a complete dataset from the trial (19). The aducanumab Phase 3 program is not the only example in the field in which interim analysis wrongly predicted futility, raising questions about the design and appropriateness of futility analyses.
Among the fundamental tenets of futility analyses is that participants included in the analysis are representative of those in the full dataset and that drop-outs are equally distributed across all treatment groups. Protocol amendments made in the course of the aducanumab study, however, resulted in non-identical interim and final populations and in cohorts that had received different doses for different periods of time
All futility analyses come with a price: loss of statistical power to demonstrate efficacy. This cost must be carefully weighed against any benefits from early termination. While there are clear advantages to stopping early when failure is inevitable, the possibility of misleading futility analyses suggests that criteria for defining failure versus success need to be very carefully specified. To implement criteria for interim analyses requires a better understanding of the clinical-biological trajectories of disease progression in stratified patient populations (19,20). Interim analyses could also benefit from looking at the totality of evidence and by aggregating signals to reduce noise.
Responder analyses could help identify subgroup differences
To determine the disease stage at which a treatment may be efficacious, the optimal duration of treatment, and other patient characteristics that may affect efficacy, responder analyses of trial data and data from open-label extension studies can be valuable. Post-hoc exploratory data analyses may yield improved understanding of study results and inform the design of future studies. For example, in the SCarlet RoAD study of gantenerumab, an exploratory analysis that classified participants according to whether they were slow or fast progressors suggested that fast progressors showed a greater exposure-dependent slowing of clinical and cognitive decline with treatment (8). While not a classic responder analysis, the exploration of the faster progressing subset allowed modeling related to a drug-placebo difference and helped to define inclusion criteria for the ongoing Phase 3 GRADUATE program with higher dose of gantenerumab.
Moving forward with amyloid-based therapies
Genetic, neuropathologic, biochemical, and now clinical trials support the amyloid hypothesis of AD while recognizing that downstream pathological processes contribute importantly to the development of the disease (6). Many questions remain to be answered in order to translate the amyloid hypothesis into efficacious therapies. For example, further research is needed to determine which Aβ species are most important to target, whether relevant Aβ species change over the course of disease, if there is an optimal time for targeting a particular Aβ species, and whether at some point amyloid becomes less relevant or irrelevant. Developing a larger repertoire of biomarkers to predict disease onset and progression, e.g. microglial activation biomarkers, may help clarify the role of amyloid-related mechanisms as well as other mechanisms in disease progression (21). Preliminary data from the monoclonal antibody trials suggest there are “downstream” effects on cerebrospinal fluid levels of neurofilament light, neurogranin, and tau. These may be crucial measures of the biological effects of interventions and that can eventually be compared across trials.
An effective treatment may also require an Aß-targeting drug in combination with a drug targeting another mechanism (e.g. neuroinflammation) or two drugs that target different amyloid mechanisms (e.g. production and clearance of Aβ) (22). Investigators have explored targeting Aß in combination with tau, the protein found in the neurofibrillary tangles that along with amyloid plaques represent the major pathological hallmarks of AD. Moving this approach forward, however, will require a better understanding of the value of various tau-related targets, the relationship of amyloid to the level of tau burden as well as the time lag between amyloid deposition, tau deposition, and cognitive impairment (23). Employing tau PET studies in clinical trials may help define these aspects of the role of tau in AD (24,25). Other tau biomarkers are in development. For example, Walsh and colleagues have shown that an N-terminal fragment of tau (NT1) and p-tau in plasma are significantly increased in AD and mild cognitive impairment (MCI) (26).
Analysis of data from several failed clinical trials of amyloid-targeting drugs suggest that to slow or prevent disease progression, it may be necessary to intervene at very early, pre-symptomatic stages of the disease (27,28). Studies currently underway to test this include the A4 study in clinically normal older individuals with elevated amyloid levels on screening PET; and the AHEAD 3-45 study in clinically normal individuals with elevated or intermediate amyloid. Other prevention trials are underway in clinically normal participants at increased genetic risk of developing AD, including the Alzheimer’s Prevention Initiative (API) Colombia Trial (20). [The DIAN-TU studies involving both clinically normal and symptomatic autosomal dominant mutation carriers recently reported negative topline results.] The challenges inherent in these prevention trials include the difficulty of detecting a slowing of progression in cognitively normal individuals and the resulting large sample size and long trial durations required; the hope of preventing AD has motivated many individuals around the world to volunteer for these studies.
Very early intervention, including primary prevention, may be more feasible with active vaccination or oral therapy rather than passive immunotherapy requiring repeated intravenous or subcutaneous administration. Active vaccination against Aß remains a plausible strategy (e.g. CAD-106; UB-311). Orally bioavailable BACE inhibitor programs have been halted with concern about observations of cognitive worsening in trials; however, evidence that this cognitive toxicity is dose-related and reversible raises hope that viable regimens may eventually move forward.
Conclusions
The termination of multiple clinical trials for futility or adverse events has had a major impact on the AD clinical research enterprise. However, evidence strongly supports amyloid as a viable target although not the only important target. Given the complexity of AD pathology, combination treatment will likely be needed. If antibody trials are sufficiently positive, they could represent a good first step towards combination treatment and lead to financial coverage and use of amyloid PET, which would be a major advance for the clinical care of AD.
To optimize the potential benefits and reduce the potential risks to participants as much as possible, methodological improvements in the design and conduct of clinical trials are needed. For example, adaptive dose finding studies may result in more patients assigned to an effective dose and avoid exposure of patients to ineffective doses. In addition, since disease modification depends on protecting neurons from the pathology, a better understanding of neuroprotection, the relationship of the biological underpinnings of the aging process (30), and the development of intermediate biomarkers of neuroprotection are needed. Advancing understanding of the complexity underlying the development of AD and potential interventions that could slow or halt the disease pathophysiological progression will require more discovery science as well as increased use of platform trials. Public-private partnerships with strong collaborations and data sharing will be necessary to accelerate these efforts, along with broad public engagement.
Acknowledgements: The authors thank Lisa J. Bain for assistance in the preparation of this manuscript.
Conflicts of interest: The Task Force was partially funded by registration fees from industrial participants. These corporations placed no restrictions on this work. Dr. Aisen reports grants from Janssen, grants from NIA, grants from FNIH, grants from Alzheimer’s Association, grants from Eisai, personal fees from Merck, personal fees from Biogen, personal fees from Roche, personal fees from Lundbeck, personal fees from Proclara, personal fees from Immunobrain Checkpoint, outside the submitted work; Dr Cummings is a consultant for Acadia, Actinogen, AgeneBio, Alkahest, Alzheon, Annovis, Avanir, Axsome, Biogen, Cassava, Cerecin, Cerevel, Cognoptix, Cortexyme, EIP Pharma, Eisai, Foresight, Gemvax, Green Valley, Grifols, Karuna, Nutricia, Orion, Otsuka, Probiodrug, ReMYND, Resverlogix, Roche, Samumed, Samus Therapeutics, Third Rock, Signant Health, Sunovion, Suven, United Neuroscience pharmaceutical and assessment companies, and the Alzheimer Drug Discovery Foundation; and owns stock in ADAMAS, BioAsis, MedAvante, QR Pharma, and United Neuroscience. Dr. Doody is an employee of Genentech/F Hoffman-LaRoche and holds stock in the company; Dr. Kramer is an employee of Eisai Company, Ltd. Dr. Kramer is an employee of Eisai Company Ltd; Dr. S. Salloway: NC; Dr. Selkoe: NC; Dr. Sims reports other from Employee of Eli Lilly and Company, outside the submitted work; Dr. Sperling reports personal fees from AC Immune, personal fees from Biogen, personal fees from Janssen, personal fees from Neurocentria , personal fees from Eisai, personal fees from GE Healthcare, personal fees from Roche, personal fees from InSightec, personal fees from Takeda Pharmaceuticals, grants from Eli Lilly , grants from Janssen, grants from Digital Cognition Technologies, grants from Eisai, grants from NIA , grants from Alzheimer’s Association, personal fees and other from Novartis, personal fees and other from AC Immune, personal fees and other from Janssen, outside the submitted work; Dr. Vellas reports grants from Lilly, Merck, Roche, Lundbeck, Biogen, grants from Alzheimer’s Association, European Commission, personal fees from Lilly, Merck, Roche, Biogen, outside the submitted work.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. Egan MF, Kost J, Voss T, Mukai Y, Aisen PS, Cummings JL, et al. Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N Engl J Med. 2019 Apr 11;380(15):1408–20.
2. Henley D, Raghavan N, Sperling R, Aisen P, Raman R, Romano G. Preliminary Results of a Trial of Atabecestat in Preclinical Alzheimer’s Disease. N Engl J Med. 2019 11;380(15):1483–5.
3. Wessels AM, Tariot PN, Zimmer JA, Selzler KJ, Bragg SM, Andersen SW, et al. Efficacy and Safety of Lanabecestat for Treatment of Early and Mild Alzheimer Disease: The AMARANTH and DAYBREAK-ALZ Randomized Clinical Trials. JAMA Neurol. 2019 Nov 25;
4. Panza F, Lozupone M, Solfrizzi V, Sardone R, Piccininni C, Dibello V, et al. BACE inhibitors in clinical development for the treatment of Alzheimer’s disease. Expert Rev Neurother. 2018 Nov;18(11):847–57.
5. Cummings JL, Cohen S, van Dyck CH, Brody M, Curtis C, Cho W, et al. ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology. 2018 22;90(21):e1889–97.
6. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016 Jun;8(6):595–608.
7. Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016 01;537(7618):50–6.
8. Ostrowitzki S, Lasser RA, Dorflinger E, Scheltens P, Barkhof F, Nikolcheva T, et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther. 2017 Dec 8;9(1):95.
9. Farlow M, Arnold SE, van Dyck CH, Aisen PS, Snider BJ, Porsteinsson AP, et al. Safety and biomarker effects of solanezumab in patients with Alzheimer’s disease. Alzheimers Dement. 2012 Jul;8(4):261–71.
10. Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014 Jan 23;370(4):311–21.
11. Siemers ER, Sundell KL, Carlson C, Case M, Sethuraman G, Liu-Seifert H, et al. Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement. 2016 Feb;12(2):110–20.
12. Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N Engl J Med. 2018 25;378(4):321–30.
13. Schwarz AJ, Sundell KL, Charil A, Case MG, Jaeger RK, Scott D, et al. Magnetic resonance imaging measures of brain atrophy from the EXPEDITION3 trial in mild Alzheimer’s disease. Alzheimers Dement (N Y). 2019;5:328–37.
14. van Dyck CH. Anti-Amyloid-β Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol Psychiatry. 2018 Feb 15;83(4):311–9.
15. Klein G, Delmar P, Voyle N, Rehal S, Hofmann C, Abi-Saab D, et al. Gantenerumab reduces amyloid-β plaques in patients with prodromal to moderate Alzheimer’s disease: a PET substudy interim analysis. Alzheimers Res Ther. 2019 Dec 12;11(1):101.
16. Willis BA, Sundell K, Lachno DR, Ferguson-Sells LR, Case MG, Holdridge K, et al. Central pharmacodynamic activity of solanezumab in mild Alzheimer’s disease dementia. Alzheimers Dement (N Y). 2018;4:652–60.
17. Panza F, Lozupone M, Seripa D, Imbimbo BP. Amyloid-β immunotherapy for alzheimer disease: Is it now a long shot? Annals of Neurology. 2019;85(3):303–15.
18. Sperling RA, Jack CR, Black SE, Frosch MP, Greenberg SM, Hyman BT, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011 Jul;7(4):367–85.
19. Doody, R. The role of futility analyses in AD clinical trials. J Prev Alz Dis. 2020;7(in press).
20. Aisen PS. Editorial: Failure After Failure. What Next in AD Drug Development? J Prev Alzheimers Dis. 2019;6(3):150.
21. Zetterberg H, Burnham SC. Blood-based molecular biomarkers for Alzheimer’s disease. Molecular Brain. 2019 Mar 28;12(1):26.
22. Gauthier S, Alam J, Fillit H, Iwatsubo T, Liu-Seifert H, Sabbagh M, et al. Combination Therapy for Alzheimer’s Disease: Perspectives of the EU/US CTAD Task Force. J Prev Alzheimers Dis. 2019;6(3):164–8.
23. Cummings J, Blennow K, Johnson K, Keeley M, Bateman RJ, Molinuevo JL, et al. Anti-Tau Trials for Alzheimer’s Disease: A Report from the EU/US/CTAD Task Force. J Prev Alzheimers Dis. 2019;6(3):157–63.
24. Harrison TM, Maass A, Adams JN, Du R, Baker SL, Jagust WJ. Tau deposition is associated with functional isolation of the hippocampus in aging. Nat Commun. 2019 Oct 25;10(1):4900.
25. Pontecorvo MJ, Devous MD, Kennedy I, Navitsky M, Lu M, Galante N, et al. A multicentre longitudinal study of flortaucipir (18F) in normal ageing, mild cognitive impairment and Alzheimer’s disease dementia. Brain. 2019 Jun 1;142(6):1723–35.
26. Chen Z, Mengel D, Keshavan A, Rissman RA, Billinton A, Perkinton M, et al. Learnings about the complexity of extracellular tau aid development of a blood-based screen for Alzheimer’s disease. Alzheimers Dement. 2019;15(3):487–96.
27. Aisen PS, Siemers E, Michelson D, Salloway S, Sampaio C, Carrillo MC, et al. What Have We Learned from Expedition III and EPOCH Trials? Perspective of the CTAD Task Force. J Prev Alzheimers Dis. 2018;5(3):171–4.
28. Sperling RA, Jack CR, Aisen PS. Testing the right target and right drug at the right stage. Sci Transl Med. 2011 Nov 30;3(111):111cm33.
29. Bateman RJ, Benzinger TL, Berry S, Clifford DB, Duggan C, Fagan AM, et al. The DIAN-TU Next Generation Alzheimer’s prevention trial: adaptive design and disease progression model. Alzheimers Dement. 2017 Jan;13(1):8–19.
30. Sierra F. Geroscience and the role of aging in the etiology and management of alzheimer’s disease. Journal of Prevention of Alzheimer’s Disease [Internet]. 2019 Mar 1 [cited 2020 Jan 30]; Available from: https://www.jpreventionalzheimer.com/all-issues.html