
R. Vassar
Corresponding Author: Ken and Ruth Davee Department of Neurology, Feinberg School of Medicine, Northwestern University, IL 60611, USA, Email: r-vassar@northwestern.edu
J Prev Alz Dis
Published online February 7, 2019, http://dx.doi.org/10.14283/jpad.2019.3
Abstract
BACE1 is the rate-limiting enzyme for the production of the Aβ peptide that forms amyloid plaques in Alzheimer’s disease (AD). Small molecule inhibitors of BACE1 are being tested in clinical trials for AD, but the safety and efficacy of BACE1 inhibition has yet to be fully explored. Knockout of the Bace1 gene in the germline of mice causes multiple neurological phenotypes, suggesting that BACE1 inhibition could be toxic. However, these phenotypes could be the result of BACE1 deficiency during development rather than due to the lack of BACE1 function in the adult. To address this problem, we generated tamoxifen-inducible conditional BACE1 knockout mice in which the Bace1 gene may be deleted in the whole body of the adult at will. Importantly, the adult conditional BACE1 knockout mice largely lack phenotypes, indicating that many BACE1 functions are not required in the adult organism. However, a germline phenotype was observed after BACE1 knockout in the adult: reduced length and disorganization of the hippocampal mossy fiber infrapyramidal bundle comprised of axons of dentate gyrus granule cells. The infrapyramidal bundle abnormality correlated with reduced proteolytic processing of the neural cell adhesion protein CHL1 that is involved in axonal guidance. We conclude that BACE1 inhibition in the adult mouse brain does not lead to the phenotypes associated with BACE1 deficiency during embryonic and postnatal development. However, adult conditional BACE1 knockout mice also suggest that BACE1 inhibitor drugs may disrupt the organization of an axonal pathway in the hippocampus, an important structure for learning and memory. Here, I review the adult conditional BACE1 knockout results and consider their implications for BACE1 inhibitor clinical trials. .
Key words: BACE1, conditional, knockout mice, axonal guidance, hippocampus, Alzheimer’s disease.
Introduction
Cerebral accumulation of amyloid-β peptide (Aβ) is a defining pathological hallmark of Alzheimer’s disease (AD) and a large body of evidence indicates that Aβ is involved in the pathogenesis of this devastating neurodegenerative disorder (1, 2). Aβ is derived from the proteolytic processing of a large type-I membrane protein, the amyloid precursor protein (APP) (3). Two proteases, the β-secretase and the γ-secretase, sequentially cut APP to generate Aβ (4). β-site APP cleaving enzyme 1 (BACE1) has been identified as the β-secretase protease that cleaves APP to initiate the production of neurotoxic Aβ (5-9). Inhibition of BACE1 and thus Aβ production has therefore emerged as a leading therapeutic intervention for AD. However, the safety of BACE1 inhibition has been questioned, because BACE1 has a wide array of substrates, and proper cleavage of these substrates may be necessary for normal physiology (10-12). Germline BACE1 knockout (BACE1-/-) mice lack Aβ production (13-15), thereby providing in vivo validation for BACE1 inhibition as a therapeutic approach for AD. However, BACE1-/- mice have been reported to exhibit smaller postnatal size and compromised survival (16), hypomyelination (17, 18), spontaneous seizures and abnormal electroencephalograms (EEGs) (19,20), memory deficits (21,22), and axon guidance defects (23-26) among other phenotypes (27). Previously, we reported that BACE1-/- mice phenocopy the axon guidance defects of CHL1 knockout mice in the hippocampus and olfactory bulb (24), indicating that BACE1 cleavage of CHL1 has a role in axonal targeting in these brain regions. However, since BACE1-/- mice are devoid of BACE1 from the moment of conception, the extent to which axon guidance defects and other BACE1-null phenotypes are related to BACE1 deficiency during development versus absence of BACE1 in the adult is unknown. This question has critical implications for the treatment of elderly AD patients with BACE1 inhibitor drugs, several of which are being tested in clinical trials (11, 28).
Generation and characterization of conditional BACE1 knockout mice
To determine the functions of BACE1 in the adult, and to model the effects of BACE1 inhibition in the mature brain, we generated conditional BACE1 knockout mice in which exon 2 of the murine BACE1 gene was flanked with loxP sites (BACE1fl/fl). This strategy enables the deletion of BACE1 exon 2 and the inactivation of the BACE1 gene following the breeding of BACE1fl/fl mice to mice that express Cre recombinase. Resulting cross-bred animals then lack the expression of BACE1 in specific spatially and temporally defined regions, tissues, or cell types. In our study (29), we crossed BACE1fl/fl mice to CamKIIα-iCre mice (30) that express Cre recombinase in early postnatal forebrain excitatory neurons (BACE1fl/fl;CamKIIα-iCre) or R26CreERT2 mice (31) that ubiquitously express from the ROSA26 locus a Cre recombinase fused to the estrogen receptor, thus enabling whole body, temporally controlled BACE1 gene deletion with administration of tamoxifen (BACE1fl/fl;R26CreERT2-TAM). For the purposes of this review, I will focus only on the results of our BACE1fl/fl;R26CreERT2-TAM conditional knockout mice, because these mice more closely model BACE1 inhibitor-treated humans in clinical trials. For those interested in the results of our BACE1fl/fl;CamKIIα-iCre mice, please refer to our recent publication in Science Translational Medicine (29).
We first verified BACE1 ablation in our conditional knockout mice by immunoblotting and immunohistochemistry. Immunostaining for BACE1 in the mouse brain showed that BACE1fl/fl;R26CreERT2mice treated with tamoxifen at 3 months and analyzed at 12 months of age exhibited little BACE1 immunostaining in cerebellum and brainstem, whereas residual BACE1 immunostaining was observed in subcortical regions and olfactory bulb (Fig. 1A). We attribute the small amount of BACE1 immunostaining in BACE1fl/fl;R26CreERT2-tamoxifen treated mice to slight variability in penetration of tamoxifen into different brain regions. Importantly, despite the low residual BACE1 immunostaining detected (in hippocampus, for example; Fig. 1A), our immunoblot analysis confirmed that BACE1fl/fl;R26CreERT2-tamoxifen treated mice exhibited robust ~90-95% BACE1 reduction in cortex and hippocampus (29).
After establishing that we were able to induce efficient adult whole-body BACE1 reduction in our conditional knockout mice, we determined the effects of conditional BACE1 gene deletion on the processing of several major BACE1 substrates: APP (5-9), close homolog of L1 (CHL1) (32,33), neuregulin 1 (NRG1) (17, 18), and seizure 6 (Sez6) (33). As expected for BACE1 substrates, in the cortex and hippocampus of BACE1fl/fl;R26CreERT2-tamoxifen-treated mice we observed increased full-length APP, CHL1, Sez6, and α-secretase-cleaved APP C-terminal fragment (α-CTF), and decreased BACE1-cleaved fragments of CHL1, NRG1, Sez6, and APP β-CTF (29). Overall, we did not observe gender differences in the effects of conditional BACE1 knockout on cleavage of BACE1 substrates.
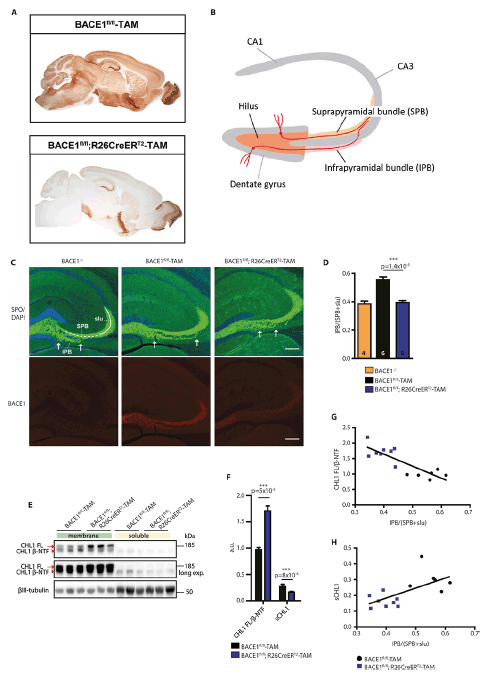
Figure 1. Adult conditional BACE1 knockout mice show disorganization of the hippocampal mossy fiber pathway
(A) DAB immunostaining for BACE1 in sagittal brain sections of BACE1fl/fl;R26CreERT2-tamoxifen treated mice compared to age-matched control BACE1fl/fl-tamoxifen treated mice. (B) Schematic diagram of the mouse hippocampus showing the dentate gyrus granule cell layer and CA1 and CA3 pyramidal cell layers in gray and the mossy fiber pathway in orange. The tracts of representative pyramidal cells in the dentate gyrus are shown in red. (C) Shown are coronal brain sections from mice with the indicated genotypes (BACE1-/-, BACE1fl/fl-TAM, BACE1fl/fl;R26CreERT2-TAM) revealing the hippocampus co-labeled for the mossy fiber marker synaptoporin (SPO, green, upper panel) and BACE1 (red, lower panel). Arrows delineate the boundaries of the infrapyramidal bundle (IPB). BACE1fl/fl-TAM and BACE1fl/fl;R26CreERT2-TAM mice were treated with tamoxifen at 3 months of age and analyzed at 1 year of age. BACE1-/- and BACE1fl/fl mice at 9 months of age were used as positive and negative controls, respectively. SPB, suprapyramidal bundle; slu, stratum lucidum. Scale bar = 200μm. (D) Infrapyramidal bundle (IPB) lengths were normalized to the lengths of the suprapyramidal bundle (SPB) plus stratum lucidum (slu) and displayed as ratios, that is, IPB/(SPB+slu). The number of mice for each genotype is indicated in each bar of the graph. Error bars indicate standard error of the mean; p-value for indicated comparison (line) using unpaired Student’s t test is shown above the bars. (E) Both soluble and membrane fractions were prepared from the hippocampi of representative BACE1fl/fl-TAM and BACE1fl/fl;R26CreERT2-TAM tamoxifen treated mice and then were subjected to immunoblot analysis for full length (FL) CHL1 and CHL1 N-terminal fragment (β-NTF). Middle panel represents a longer exposure of the upper panel. βIII-tubulin was the loading control. (F) Ratio of CHL1 FL to CHL1 β-NTF intensities in membrane fractions (CHL1 FL/ β-NTF) and the intensity of the CHL1 β-NTF band in soluble fractions normalized to βIII-tubulin (sCHL1) displayed as arbitrary units (a.u.) BACE1fl/fl ;R26CreERT2-TAM and BACE1fl/fl-TAM mice treated with tamoxifen, mean ± SEM. (G) Correlation of CHL1 FL/ β-NTF from (F) plotted against IPB/(SPB+slu) for BACE1fl/fl;R26CreERT2-TAM and BACE1fl/fl-TAM tamoxifen treated mice (R2=0.7014, p=0.0002). (H) Correlation of sCHL1 from (F) plotted against IPB/(SPB+slu) for BACE1fl/fl;R26CreERT2-TAM and BACE1fl/fl-TAM tamoxifen treated mice (R2=0.4117, p=0.01).
Conditional BACE1 knockout mice are cognitively normal, but exhibit signs of hyperactivity
We next evaluated learning and memory function in our conditional BACE1 knockout mice, as BACE1-/- mice were previously reported to have memory deficits as early as 3 months of age (21, 22). BACE1fl/fl;R26CreERT2 mice treated with tamoxifen at 3 months of age and assessed at 9 months of age (6 months following Bace1 gene deletion) performed comparably to BACE1fl/fl-tamoxifen treated mice in the Morris water maze, spontaneous alternation in the Y-maze, and contextual and cued fear conditioning (29). Hippocampal CA1 longterm potentiation (LTP) of the BACE1fl/fl;R26CreERT2-tamoxifen treated mice was also normal. We assessed locomotor activity of the mice in the open field test and while there were no differences in basal motor activity for conditional knockout mice, we observed greater arm entries in the Y-maze, but did not show longer travel distance in the open field test. Together, these results suggest that although conditional Bace1 gene inactivation may be associated with mild hyperactivity, it does not cause memory deficits if it occurs in the adult.
Adult whole-body conditional BACE1 knockout mice lack epileptiform abnormalities and hypomyelination
Spontaneous seizure and abnormal EEGs are other adverse phenotypes that have been reported in BACE1-/- mice (19, 20). To assess seizure activity in our conditional BACE1 knockout mice, we counted the number of spontaneous seizures and subjected mice to EEG recording and video monitoring. Importantly, we observed no spontaneous seizures in 86 BACE1fl/fl;R26CreERT2-tamoxifen treated and 62 BACE1fl/fl-tamoxifen treated mice (29). EEG recordings from BACE1fl/fl;R26CreERT2 mice treated with tamoxifen at 3 months and analyzed at 1 year of age (9 months following Bace1 gene deletion) showed no seizures or abnormal EEG activity compared to control BACE1fl/fl-tamoxifen treated mice. These results suggest that spontaneous seizures and abnormal EEGs derive from BACE1 deficiency during development, but that BACE1 conditional knockout in adult neurons may not lead to epileptiform activity.
BACE1-/- mice were reported to have hypomyelination from insufficient BACE1 cleavage of NRG1 and impaired downstream signaling (17, 18). To determine whether our BACE1 conditional knockout mice also exhibited hypomyelination, we performed immunoblot analysis of cortex and sciatic nerve homogenates for myelin basic protein (MBP) and myelin proteolipid protein (PLP), and stained brain sections with Luxol fast blue for white matter. Immunoblot analysis revealed no difference in central nervous system (CNS) myelination in BACE1fl/fl;R26CreERT2 mice treated with tamoxifen at 3 months and analyzed at 12 months (9 months following Bace1 gene deletion) either by cortical or hippocampal MBP immunoblotting or Luxol fast blue staining of brain sections (29). Similarly, while immunoblot analysis of sciatic nerve homogenates from BACE1fl/fl;R26CreERT2-tamoxifen treated mice showed undetectable BACE1, we found no change in MBP compared to BACE1fl/fl-tamoxifen treated control or BACE1+/+ mice, even though BACE1-/- sciatic nerve homogenates displayed a significant reduction of MBP. We further measured myelin thickness and calculated g-ratios in semi-thin sciatic nerve sections of BACE1fl/fl;R26CreERT2- tamoxifen treated mice along with BACE1fl/fl- tamoxifen treated, BACE-/- and BACE1+/+ control animals. Sciatic nerve of BACE1-/- mice, as previously reported (17,18), exhibited reduced myelin sheath thickness around axons and significantly increased g-ratio indicating hypomyelination. In contrast, BACE1fl/fl;R26CreERT2-tamoxifen treated mice displayed normal myelin sheath thickness and a g-ratio similar to BACE1fl/fl-tamoxifen treated mice and BACE1+/+ mice. Thus, although germline Bace1 gene deletion causes hypomyelination, BACE1 conditional knockout in the adult whole body does not seem to affect central or peripheral myelination once development is completed.
Adult conditional BACE1 knockout mice exhibit axonal organization defects in the hippocampus
Previously, we determined that BACE1-/- mice exhibited axonal organization defects that correlated with deficient processing of the neural cell adhesion molecule CHL1 (24). CHL1 is a substrate that appears to be preferred by BACE1 in neurons (32,33). Consistent with these observations, BACE1-cleaved CHL1 β-NTF and full length CHL1 were decreased and increased, respectively, in the brains of BACE1fl/fl;R26CreERT2-tamoxifen treated mice (29). Interestingly, differences in CHL1 β-NTF and full length CHL1 between BACE1fl/fl;R26CreERT2-tamoxifen treated mice and BACE1fl/fl-tamoxifen treated control mice were substantial, suggesting that induced deletion of the Bace1 gene in the adult may affect the function of CHL1 even after development is completed.
One of the CHL1-correlated axonal abnormalities that we had observed in the brains of germline BACE1 knockout mice was a disorganized and shortened length of the infrapyramidal bundle (IPB) of the hippocampal mossy fiber pathway and premature crossing of the CA3 pyramidal cell layer (24). The IPB consists of the set of axons that project from granule cells of the lower blade of the dentate gyrus that run below the pyramidal cell layer of CA3 (Fig. 1B). The IPB extends for a distance distal to the dentate gyrus and then traverses the pyramidal cell layer to join axons of the suprapyramidal bundle (SPB). In mice, the length of the IPB positively correlates with fewer errors in the radial maze, indicating increased spatial learning capabilities (34). Additionally, training mice in the Morris water maze and induction of LTP increases IPB size, suggesting spatial learning induces structural plasticity of the IPB (35). Given the role of the dentate gyrus in pattern separation (36), the length of the IPB may have a significance impact on spatial learning and memory.
To determine whether deficient BACE1 cleavage of CHL1 correlated with axonal defects in adult mice, we measured the length and assessed the organization of the IPB of BACE1fl/fl;R26CreERT2-tamoxifen treated mice in coronal brain sections immunostained for BACE1 and the mossy fiber marker synaptoporin (29). Similar to BACE1-/- mice, IPB length was shorter in BACE1fl/fl;R26CreERT2 mice treated with tamoxifen at 3 months and analyzed at 1 year of age (9 months following Bace1 gene deletion) compared to BACE1fl/fl-tamoxifen treated negative control mice (Fig. 1C, D). The amount that the IPB was shortened in the adult conditional knockout mice (~30%) was similar to that observed in BACE1-/- mice reported in our previous study (24). Also similar to BACE1-/- mice, we observed that the organization of the IPB of BACE1fl/fl;R26CreERT2-tamoxifen treated mice was severely disrupted compared to that of BACE1fl/fl-tamoxifen treated mice, exhibiting premature crossing of mossy fibers through the CA3 pyramidal cell layer (Fig. 1C); this is a phenocopy of CHL1 knockout mice (37, 38). Immunostaining with an anti-CHL1 antibody revealed co-localization of CHL1 with presynaptic terminal and mossy fiber bouton markers synaptophysin and calbindin, respectively, indicating neuronal, and specifically presynaptic, CHL1 localization. As we have previously reported, CHL1 also co-localized with BACE1 in mossy fiber terminals in the hippocampus (24).
Next, we performed immunoblot analysis for CHL1 to determine the extent of BACE1 cleavage of CHL1 in the hippocampus of the adult conditional BACE1 knockout and control mice. As expected, we found that CHL1 β-NTF and full length CHL1 were decreased and increased in soluble and membrane fractions of hippocampal homogenates, respectively, in BACE1fl/fl;R26CreERT2-tamoxifen treated mice compared to BACE1fl/fl-tamoxifen treated mice (Fig. 1E, F). Linear regression analysis showed that IPB length positively and negatively correlated with soluble CHL1 and the ratio of full length CHL1 to β-NTF, respectively (Fig. 1G, H). Finally, we used BrdU labeling experiments to determine that the axonal organization defects in the IPB of adult conditional BACE1 knockout mice were not associated with abnormal neural progenitor cell proliferation, adult neurogenesis, or cell death. Taken together, our results demonstrate that processing of CHL1 by BACE1 is active in the adult brain and is correlated with the maintenance of the normal structure of a major axonal pathway in the hippocampus that is involved in learning and memory.
Implications of conditional BACE1 knockout mice for the role of BACE1 in the adult brain
We generated conditional BACE1 knockout mice to determine whether BACE1 is required in the adult for normal brain structure and function. Several of the neurological phenotypes of germline BACE1 knockout mice were absent following adult conditional BACE1 gene deletion (Table 1). Importantly, however, when we allowed the brain to develop with BACE1 and then ablated BACE1 in the adult, we observed that the IPB of the hippocampal mossy fiber pathway was disorganized and exhibited a shortened length, possibly as the result of a defect in axon guidance. BACE1 is highly concentrated in hippocampal mossy fibers (39,40), perhaps for the continuing requirement of adult axon guidance (41, 42). Indeed, CHL1 is a BACE1 substrate (32, 33), and we have shown that BACE1-/- mice phenocopy the axon organization defects of CHL1 knockout mice (24). Moreover, the cleavage of CHL1 by BACE1 regulates the balance between growth cone extension and collapse via the axon guidance protein semaphorin 3A (26), a process critical for correct axonal targeting. Future studies are necessary to determine whether axonal disorganization exists beyond the hippocampus in other regions of the brain of adult conditional BACE1 knockout mice. Additionally, treatment of wild-type mice with a BACE1 inhibitor should be performed to determine whether IPB organizational defects occur following pharmacological BACE1 inhibition.
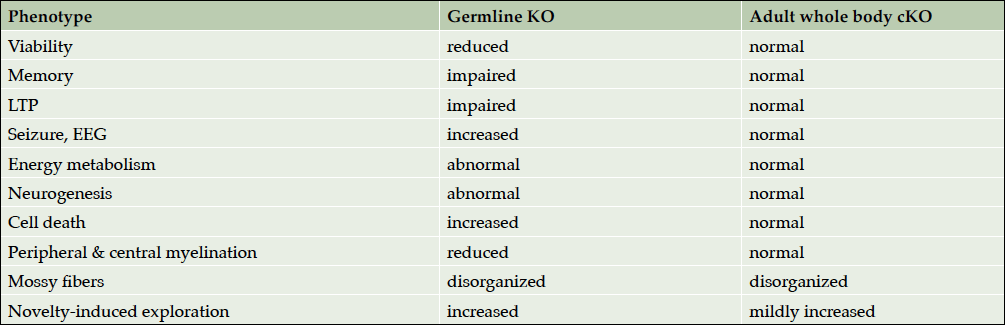
Table 1. Summary of adult conditional BACE1 knockout phenotypes
Neurogenesis of dentate gyrus granule cells is known to continue in the subgranular zone of the hippocampus during adulthood (43). Consequently, mossy fiber axons of granule cells born in the adult require axon guidance to find their correct target neurons in the hippocampal CA3 region. Mossy fiber axons of newly-born granule cells in the infrapyramidal blade of the dentate gyrus grow for a distance along the ventral side of the CA3 pyramidal cell layer, thus forming the IPB. The axons then invade the cell layer and cross to the dorsal side where they merge into the stratum lucidum and join other mossy fiber axons that come from the suprapyramidal blade of the dentate gyrus. The factors that determine the distance that mossy fibers grow before crossing the CA3 pyramidal cell layer, and therefore the length of the IPB, are not yet fully elucidated, but correlate with CHL1 processing by BACE1 (24).
Interestingly, the length of the IPB varies between different inbred strains of mice, and longer IPB lengths correlate with better spatial orientation learning in the radial maze (44). Given this finding, it was unexpected that we did not observe memory deficits in adult conditional BACE1 knockout mice in our study. In future studies, it will be important to test adult conditional BACE1 knockout mice in more sensitive and varied behavioral paradigms, including the radial maze and Barnes maze that avoid the stress of swimming, to detect potential deficits in spatial orientation and other types of learning or memory with age. Moreover, germline BACE1 knockout mice have been reported to exhibit impaired mossy fiber-CA3 LTP (45,46). Therefore, it will be important to determine whether adult conditional BACE1 knockout mice have a mossy fiber-CA3 LTP abnormality and, if so, understand its relationship to the IPB and behavioral phenotypes.
Although we favor the hypothesis that deficient BACE1 cleavage of CHL1 causes defective axon guidance resulting in the short and disorganized IPB of adult conditional knockout mice, other mechanisms, either involving CHL1 or other BACE1 substrates such as Sez6 or NRG1, could have a role. Additionally, other mechanisms besides axon guidance may contribute to the abnormal IPB associated with BACE1 deficiency, including synaptic maintenance or plasticity. Our BrdU experiments indicated that alterations in adult neurogenesis including overall or regional neural progenitor cell proliferation, neuronal differentiation, and cell death in the dentate gyrus were unlikely to produce the IPB phenotype. However, we could not exclude other potential causes, such as abnormal neurite outgrowth or defective activity dependent axonal refining, among others.
It is possible that a combination of mechanisms may influence the length and organization of the IPB. For example, impaired axon guidance together with axonal degeneration as observed in germline BACE1 knockout mice (19) may affect the IPB. A study reported that only ~10% of dentate gyrus neurons are newly generated by adult neurogenesis in mice, although the authors noted that this number is an underestimate (47). The same authors also observed that relatively few dentate gyrus neurons die, and that the total number of granule cells continues to increase in the adult dentate gyrus. These results suggest that defective axon guidance of newly generated mossy fibers together with degeneration of axons formed during development may contribute to the shortened and disorganized IPB of adult conditional BACE1 knockout mice. Since BACE1 cleavage of CHL1 regulates growth cone collapse (26), it is possible that both axon guidance and mossy fiber degeneration are affected by CHL1 processing in the adult dentate gyrus. It will be important to conduct additional studies to investigate the potential roles of these and other mechanisms in determining the characteristics of the IPB.
Implications of axonal organization defects in adult conditional BACE1 knockout mice for BACE1 inhibitor clinical trials
Adult conditional BACE1 knockout mice are protected from adverse phenotypes found in germline BACE1 null mice, including reduced survival, growth retardation, seizures, EEG abnormalities, hypomyelination, and memory deficits (Table 1). We conclude that most germline BACE1 knockout mouse phenotypes are the result of BACE1 deficiency during development, not from the absence of BACE1 function in the adult. This conclusion is also supported by the observation that BACE1 and many of its substrates are highly expressed in the developing organism, especially in the nervous system (17,18). On the one hand, our results imply that BACE1 inhibitor treatment in adults may be safe for the most part. On the other hand, the fact that adult conditional BACE1 knockout mice exhibit aberrant morphology in an axonal pathway important for learning and memory suggests that BACE1 inhibition may not be free of mechanism-based side effects in the adult brain. Indeed, as was recently reported at the 11th Clinical Trials on Alzheimer’s Conference in Barcelona, Spain, compared to placebo BACE1 inhibitor treatment resulted in dose-dependent cognitive worsening, neuropsychiatric disturbances, and hippocampal volume loss in Phase 2 and 3 studies in subjects with prodromal or early AD, despite significant lowering of Aβ in the CNS. Although these effects were small and not observed for all BACE1 inhibitors, they occurred for four individual drugs, suggesting on-target class issues rather than compound-specific effects. Only two BACE1 inhibitor clinical trials are currently ongoing, the rest having been discontinued before completion.
The mechanism(s) of the cognitive worsening, neuropsychiatric disturbances, and hippocampal volume loss observed in the BACE1 inhibitor clinical trials has yet to be determined. Effects occurred soon after the start of treatment but stabilized and did not increase over time, suggesting that that they were not the result of an accelerated neurodegenerative process. These characteristics indicate a synaptic mechanism, consistent with the presynaptic localization of BACE1 and many of its substrates. The BACE1 inhibitor trials were designed to reduce Aβ levels in the CNS by ~60-90%, which represents strong BACE1 inhibition and is on par with the level of inhibition achieved in our adult conditional BACE1 knockout mice. It is tempting to speculate that the adverse effects reported in the human trials are related to axonal organization defects caused by strongly inhibited BACE1 cleavage of CHL1, as demonstrated in the adult conditional BACE1 knockout mice. However, other mechanisms involving different BACE1 substrates that affect synaptic plasticity or maintenance could also have a role. Further investigations of adult conditional BACE1 knockout mice may provide additional insights into this important question.
Although it is too soon to draw firm conclusions about the ultimate safety of BACE1 inhibition, it is possible that the toxicities observed in the current clinical trials may have been the result of BACE1 over-inhibition. The APP A673T mutation is associated with protection against AD (48) and lowers Aβ by only a modest amount, ~28% (49). Additionally, germline heterozygous BACE1 knockout mice with 50% of the normal BACE1 level in the brain are indistinguishable from wild-type mice (13,21,50). Together, these observations suggest that a BACE1 inhibitor dose that achieves up to 50% inhibition should be both safe and effective if administered at an early stage of disease when amyloid pathology is low. Similar to the paradigm of the cholesterol-lowering statin drugs for the prevention of heart disease, low doses of BACE1 inhibitors may be safe and effective for the prevention of Alzheimer’s disease if administered presymtomatically.
Acknowledgements: I thank members of the Vassar lab for helpful discussions and assistance, especially Dr. Shahrnaz Kemal for generating the figure.
Funding: This work was supported by NIH R01 AG022560, BrightFocus Foundation, and the Baila Foundation.
Conflict of interest: R. Vassar is a consultant for Eisai, Lilly, and Novartis, and is on the Scientific Advisory Board of Alector.
Ethical standards: The study protocol was approved by the Northwestern University Institutional Animal Care and Use Committee.
Open Access: This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
References
1. Tanzi, R. E. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med 2, 2012
2. Selkoe, D. J., and Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO molecular medicine 2016;8, 595-608
3. Tanzi, R. E., Gusella, J. F., Watkins, P. C., Bruns, G. A., St George-Hyslop, P., Van Keuren, M. L., Patterson, D., Pagan, S., Kurnit, D. M., and Neve, R. L. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science 1987;235, 880-884
4. Haass, C., Kaether, C., Thinakaran, G., and Sisodia, S. (2012) Trafficking and proteolytic processing of APP. Cold Spring Harbor perspectives in medicine 2012;2, a006270
5. Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., Teplow, D. B., Ross, S., Amarante, P., Loeloff, R., Luo, Y., Fisher, S., Fuller, J., Edenson, S., Lile, J., Jarosinski, M. A., Biere, A. L., Curran, E., Burgess, T., Louis, J. C., Collins, F., Treanor, J., Rogers, G., and Citron, M. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999;286, 735-741
6. Yan, R., Bienkowski, M. J., Shuck, M. E., Miao, H., Tory, M. C., Pauley, A. M., Brashier, J. R., Stratman, N. C., Mathews, W. R., Buhl, A. E., Carter, D. B., Tomasselli, A. G., Parodi, L. A., Heinrikson, R. L., and Gurney, M. E. Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature 1999;402, 533-537
7. Sinha, S., Anderson, J. P., Barbour, R., Basi, G. S., Caccavello, R., Davis, D., Doan, M., Dovey, H. F., Frigon, N., Hong, J., Jacobson-Croak, K., Jewett, N., Keim, P., Knops, J., Lieberburg, I., Power, M., Tan, H., Tatsuno, G., Tung, J., Schenk, D., Seubert, P., Suomensaari, S. M., Wang, S., Walker, D., Zhao, J., McConlogue, L., and John, V. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 1999;402, 537-540
8. Hussain, I., Powell, D., Howlett, D. R., Tew, D. G., Meek, T. D., Chapman, C., Gloger, I. S., Murphy, K. E., Southan, C. D., Ryan, D. M., Smith, T. S., Simmons, D. L., Walsh, F. S., Dingwall, C., and Christie, G. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci 1999;14, 419-427
9. Lin, X., Koelsch, G., Wu, S., Downs, D., Dashti, A., and Tang, J. Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc Natl Acad Sci U S A 2000;97, 1456-1460
10. Barao, S., Moechars, D., Lichtenthaler, S. F., and De Strooper, B. BACE1 Physiological Functions May Limit Its Use as Therapeutic Target for Alzheimer’s Disease. Trends in neurosciences 2016;39, 158-169
11. Yan, R., and Vassar, R. Targeting the beta secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol 2014;13, 319-329
12. Yan, R. Physiological Functions of the beta-Site Amyloid Precursor Protein Cleaving Enzyme 1 and 2. Frontiers in molecular neuroscience 201710, 97
13. Luo, Y., Bolon, B., Kahn, S., Bennett, B. D., Babu-Khan, S., Denis, P., Fan, W., Kha, H., Zhang, J., Gong, Y., Martin, L., Louis, J. C., Yan, Q., Richards, W. G., Citron, M., and Vassar, R. Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci 2001;4, 231-232
14. Cai, H., Wang, Y., McCarthy, D., Wen, H., Borchelt, D. R., Price, D. L., and Wong, P. C. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci 2001;4, 233-234
15. Roberds, S. L., Anderson, J., Basi, G., Bienkowski, M. J., Branstetter, D. G., Chen, K. S., Freedman, S. B., Frigon, N. L., Games, D., Hu, K., Johnson-Wood, K., Kappenman, K. E., Kawabe, T. T., Kola, I., Kuehn, R., Lee, M., Liu, W., Motter, R., Nichols, N. F., Power, M., Robertson, D. W., Schenk, D., Schoor, M., Shopp, G. M., Shuck, M. E., Sinha, S., Svensson, K. A., Tatsuno, G., Tintrup, H., Wijsman, J., Wright, S., and McConlogue, L. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer’s disease therapeutics. Human molecular genetics 2001;10, 1317-1324
16. Dominguez, D., Tournoy, J., Hartmann, D., Huth, T., Cryns, K., Deforce, S., Serneels, L., Camacho, I. E., Marjaux, E., Craessaerts, K., Roebroek, A. J., Schwake, M., D’Hooge, R., Bach, P., Kalinke, U., Moechars, D., Alzheimer, C., Reiss, K., Saftig, P., and De Strooper, B. Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J Biol Chem 2005;280, 30797-30806
17. Willem, M., Garratt, A. N., Novak, B., Citron, M., Kaufmann, S., Rittger, A., DeStrooper, B., Saftig, P., Birchmeier, C., and Haass, C. Control of peripheral nerve myelination by the beta-secretase BACE1. Science 2006;314, 664-666
18. Hu, X., Hicks, C. W., He, W., Wong, P., Macklin, W. B., Trapp, B. D., and Yan, R. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci 2006;9, 1520-1525
19. Hu, X., Zhou, X., He, W., Yang, J., Xiong, W., Wong, P., Wilson, C. G., and Yan, R. BACE1 deficiency causes altered neuronal activity and neurodegeneration. J Neurosci 2010;30, 8819-8829
20. Hitt, B. D., Jaramillo, T. C., Chetkovich, D. M., and Vassar, R. BACE1-/- mice exhibit seizure activity that does not correlate with sodium channel level or axonal localization. Mol Neurodegener 2010;5, 31
21. Laird, F. M., Cai, H., Savonenko, A. V., Farah, M. H., He, K., Melnikova, T., Wen, H., Chiang, H. C., Xu, G., Koliatsos, V. E., Borchelt, D. R., Price, D. L., Lee, H. K., and Wong, P. C. BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci 2005;25, 11693-11709
22. Ohno, M., Chang, L., Tseng, W., Oakley, H., Citron, M., Klein, W. L., Vassar, R., and Disterhoft, J. F. Temporal memory deficits in Alzheimer’s mouse models: rescue by genetic deletion of BACE1. Eur J Neurosci 2006;23, 251-260
23. Rajapaksha, T. W., Eimer, W. A., Bozza, T. C., and Vassar, R. The Alzheimer’s beta-secretase enzyme BACE1 is required for accurate axon guidance of olfactory sensory neurons and normal glomerulus formation in the olfactory bulb. Mol Neurodegener 2011;6, 88
24. Hitt, B., Riordan, S. M., Kukreja, L., Eimer, W. A., Rajapaksha, T. W., and Vassar, R. beta-Site amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1)-deficient mice exhibit a close homolog of L1 (CHL1) loss-of-function phenotype involving axon guidance defects. The Journal of biological chemistry 2012;287, 38408-38425
25. Cao, L., Rickenbacher, G. T., Rodriguez, S., Moulia, T. W., and Albers, M. W. The precision of axon targeting of mouse olfactory sensory neurons requires the BACE1 protease. Sci Rep 2012;2, 231
26. Barao, S., Gartner, A., Leyva-Diaz, E., Demyanenko, G., Munck, S., Vanhoutvin, T., Zhou, L., Schachner, M., Lopez-Bendito, G., Maness, P. F., and De Strooper, B. Antagonistic Effects of BACE1 and APH1B-gamma-Secretase Control Axonal Guidance by Regulating Growth Cone Collapse. Cell reports 2015;12, 1367-1376
27. Vassar, R., Kovacs, D. M., Yan, R., and Wong, P. C. The beta-secretase enzyme BACE in health and Alzheimer’s disease: regulation, cell biology, function, and therapeutic potential. J Neurosci 2009;29, 12787-12794
28. Yan, R. Stepping closer to treating Alzheimer’s disease patients with BACE1 inhibitor drugs. Translational neurodegeneration 20165, 13
29. Ou-Yang, M. H., Kurz, J. E., Nomura, T., Popovic, J., Rajapaksha, T. W., Dong, H., Contractor, A., Chetkovich, D. M., Tourtellotte, W. G., and Vassar, R. Axonal organization defects in the hippocampus of adult conditional BACE1 knockout mice. Science translational medicine 2018;10
30. Casanova, E., Fehsenfeld, S., Mantamadiotis, T., Lemberger, T., Greiner, E., Stewart, A. F., and Schutz, G. A CamKIIalpha iCre BAC allows brain-specific gene inactivation. Genesis 2001;31, 37-42
31. Ventura, A., Kirsch, D. G., McLaughlin, M. E., Tuveson, D. A., Grimm, J., Lintault, L., Newman, J., Reczek, E. E., Weissleder, R., and Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007;445, 661-665
32. Zhou, L., Barao, S., Laga, M., Bockstael, K., Borgers, M., Gijsen, H., Annaert, W., Moechars, D., Mercken, M., Gevaert, K., and De Strooper, B. The neural cell adhesion molecules L1 and CHL1 are cleaved by BACE1 protease in vivo. The Journal of biological chemistry 2012;287, 25927-25940
33. Kuhn, P. H., Koroniak, K., Hogl, S., Colombo, A., Zeitschel, U., Willem, M., Volbracht, C., Schepers, U., Imhof, A., Hoffmeister, A., Haass, C., Rossner, S., Brase, S., and Lichtenthaler, S. F. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J 2012;31, 3157-3168
34. Schwegler, H., Crusio, W. E., and Brust, I. Hippocampal mossy fibers and radial-maze learning in the mouse: a correlation with spatial working memory but not with non-spatial reference memory. Neuroscience 1990;34, 293-298
35. Wiera, G., and Mozrzymas, J. W. Extracellular proteolysis in structural and functional plasticity of mossy fiber synapses in hippocampus. Frontiers in cellular neuroscience 2015;9, 427
36. Schmidt, B., Marrone, D. F., and Markus, E. J. Disambiguating the similar: the dentate gyrus and pattern separation. Behav Brain Res 2012;226, 56-65
37. Montag-Sallaz, M., Schachner, M., and Montag, D. Misguided axonal projections, neural cell adhesion molecule 180 mRNA upregulation, and altered behavior in mice deficient for the close homolog of L1. Mol Cell Biol 2002;22, 7967-7981
38. Heyden, A., Angenstein, F., Sallaz, M., Seidenbecher, C., and Montag, D. Abnormal axonal guidance and brain anatomy in mouse mutants for the cell recognition molecules close homolog of L1 and NgCAM-related cell adhesion molecule. Neuroscience 2008;155, 221-233
39. Kandalepas, P. C., Sadleir, K. R., Eimer, W. A., Zhao, J., Nicholson, D. A., and Vassar, R. The Alzheimer’s beta-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol 2013;126, 329-352
40. Zhao, J., Fu, Y., Yasvoina, M., Shao, P., Hitt, B., O’Connor, T., Logan, S., Maus, E., Citron, M., Berry, R., Binder, L., and Vassar, R. Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer’s disease pathogenesis. J Neurosci 2007;27, 3639-3649
41. Faulkner, R. L., Jang, M. H., Liu, X. B., Duan, X., Sailor, K. A., Kim, J. Y., Ge, S., Jones, E. G., Ming, G. L., Song, H., and Cheng, H. J. Development of hippocampal mossy fiber synaptic outputs by new neurons in the adult brain. Proceedings of the National Academy of Sciences of the United States of America 2008;105, 14157-14162
42. Toni, N., Laplagne, D. A., Zhao, C., Lombardi, G., Ribak, C. E., Gage, F. H., and Schinder, A. F. Neurons born in the adult dentate gyrus form functional synapses with target cells. Nature neuroscience 2008;11, 901-907
43. Goncalves, J. T., Schafer, S. T., and Gage, F. H. (2016) Adult Neurogenesis in the Hippocampus: From Stem Cells to Behavior. Cell 2016;167, 897-914
44. Crusio, W. E., and Schwegler, H. Learning spatial orientation tasks in the radial-maze and structural variation in the hippocampus in inbred mice. Behav Brain Funct 2005;1, 3
45. Wang, H., Song, L., Laird, F., Wong, P. C., and Lee, H. K. BACE1 knock-outs display deficits in activity-dependent potentiation of synaptic transmission at mossy fiber to CA3 synapses in the hippocampus. J Neurosci 2008;28, 8677-8681
46. Wang, H., Song, L., Lee, A., Laird, F., Wong, P. C., and Lee, H. K. Mossy fiber long-term potentiation deficits in BACE1 knock-outs can be rescued by activation of alpha7 nicotinic acetylcholine receptors. The Journal of neuroscience : the official journal of the Society for Neuroscience 2010;30, 13808-13813
47. Imayoshi, I., Sakamoto, M., Ohtsuka, T., Takao, K., Miyakawa, T., Yamaguchi, M., Mori, K., Ikeda, T., Itohara, S., and Kageyama, R. Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nature neuroscience 2008;11, 1153-1161
48. Jonsson, T., Atwal, J. K., Steinberg, S., Snaedal, J., Jonsson, P. V., Bjornsson, S., Stefansson, H., Sulem, P., Gudbjartsson, D., Maloney, J., Hoyte, K., Gustafson, A., Liu, Y., Lu, Y., Bhangale, T., Graham, R. R., Huttenlocher, J., Bjornsdottir, G., Andreassen, O. A., Jonsson, E. G., Palotie, A., Behrens, T. W., Magnusson, O. T., Kong, A., Thorsteinsdottir, U., Watts, R. J., and Stefansson, K. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012;488, 96-99
49. Martiskainen, H., Herukka, S. K., Stancakova, A., Paananen, J., Soininen, H., Kuusisto, J., Laakso, M., and Hiltunen, M. Decreased plasma beta-amyloid in the Alzheimer’s disease APP A673T variant carriers. Ann Neurol 2017;82, 128-132
50. Roberds, S. L., Anderson, J., Basi, G., Bienkowski, M. J., Branstetter, D. G., Chen, K. S., Freedman, S. B., Frigon, N. L., Games, D., Hu, K., Johnson-Wood, K., Kappenman, K. E., Kawabe, T. T., Kola, I., Kuehn, R., Lee, M., Liu, W., Motter, R., Nichols, N. F., Power, M., Robertson, D. W., Schenk, D., Schoor, M., Shopp, G. M., Shuck, M. E., Sinha, S., Svensson, K. A., Tatsuno, G., Tintrup, H., Wijsman, J., Wright, S., and McConlogue, L. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer’s disease therapeutics. Hum. Mol. Genet. 2001;10, 1317-1324